How to Run FastQC and MultiQC on Raw RNA-Seq Reads
The most expensive mistake in RNA-seq is running a full alignment pipeline on data you never should have aligned. A sample with a RIN of 4, 40% adapter contamination, or a mapping rate that tells you the wrong organism got sequenced — none of these problems are fixable downstream. They are only detectable if you actually look at your QC output before moving on.
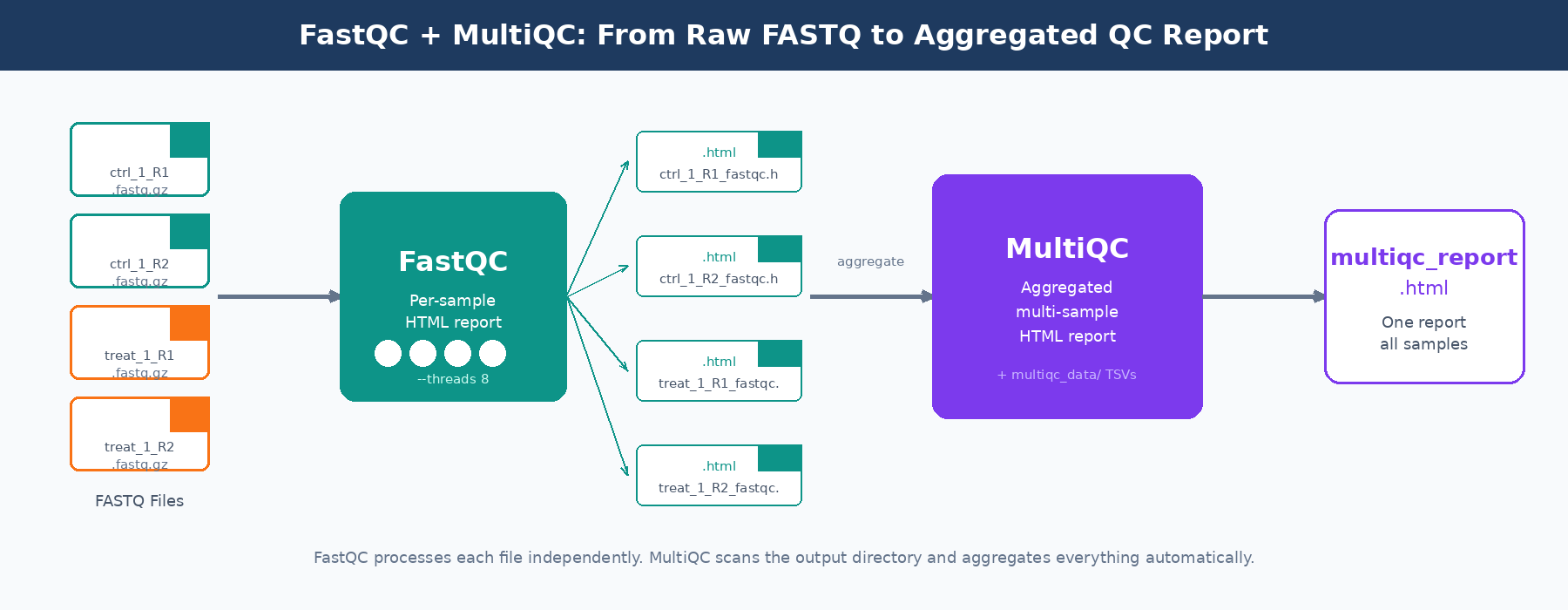
FastQC and MultiQC are the standard tools for this. FastQC produces a per-sample HTML report covering a dozen quality metrics. MultiQC aggregates those reports across all your samples into a single interactive HTML file, making cross-sample outlier detection practical. Together they take about ten minutes to run on a 30-sample experiment and save you hours of debugging bad alignments.
This tutorial walks through installation, running both tools on real data, automating the process across large sample sets, and parsing MultiQC’s machine-readable output in Python to programmatically flag failing samples. If you want the broader pipeline context around where QC sits, read Raw Reads to Counts: The Bulk RNA-Seq Pipeline Explained. If your FastQC report suggests adapter contamination, continue with Trimming Adapters with Trimmomatic and fastp: A Side-by-Side Walkthrough.
Installation
Both tools are available through conda, which is the cleanest way to manage bioinformatics software without fighting system-level dependencies.
# Create a dedicated environment for QC toolsconda create -n rnaseq-qc -c bioconda -c conda-forge fastqc multiqc -y
# Activate itconda activate rnaseq-qc
# Verify versionsfastqc --versionmultiqc --versionIf you are not using conda, FastQC can be installed via sudo apt install fastqc on Ubuntu-based systems. MultiQC is a Python package and installs cleanly with pip install multiqc. It requires Python 3.7 or later.
On an HPC cluster, both are almost certainly available as modules. Check with module spider fastqc and load the appropriate version before running batch jobs.
Running FastQC on a Single Sample
FastQC’s interface is straightforward. For one paired-end sample:
# Create output directorymkdir -p results/fastqc
# Run on both read files of a paired-end samplefastqc \ --outdir results/fastqc \ --threads 4 \ data/raw/sample_ctrl_1_R1.fastq.gz \ data/raw/sample_ctrl_1_R2.fastq.gzFastQC produces two files per input: an HTML report and a ZIP archive. The ZIP contains a fastqc_data.txt file with all the numeric data underlying the plots, and a summary.txt with pass/warn/fail flags per module. You generally open the HTML to look at plots, and use the ZIP contents when you want to parse results programmatically.
The --threads flag controls how many files FastQC processes in parallel. It does not speed up processing of a single file. For a batch of 20 files, set --threads 20 and FastQC will process them simultaneously, dramatically cutting wall time.
FastQC flags are tuned for WGS, not RNA-seq
FastQC’s pass/warn/fail thresholds are calibrated for whole-genome shotgun data. For RNA-seq, “warn” on per-base sequence content in the first 12 bases is normal and caused by random hexamer priming bias. “Fail” on sequence duplication is expected for highly expressed genes. Read the numbers, not just the color-coded flags.
Automating FastQC Across Many Samples
Running FastQC one sample at a time on a 40-sample experiment is not practical. Here is a bash script that processes all paired-end FASTQ files in a directory in parallel, using a job array pattern suitable for both local machines and HPC environments.
#!/usr/bin/env bash# Usage: bash run_fastqc.sh /path/to/fastq/dir /path/to/output/dir
FASTQ_DIR="${1}"OUT_DIR="${2:-results/fastqc}"THREADS=8
mkdir -p "${OUT_DIR}"
# Find all R1 files and derive R2 pathsmapfile -t r1_files < <(find "${FASTQ_DIR}" -name "*_R1*.fastq.gz" | sort)
echo "Found ${#r1_files[@]} sample(s). Running FastQC..."
for r1 in "${r1_files[@]}"; do # Derive R2 path by substituting _R1 with _R2 r2="${r1/_R1/_R2}"
if [[ -f "${r2}" ]]; then fastqc \ --outdir "${OUT_DIR}" \ --threads 2 \ --quiet \ "${r1}" "${r2}" & else # Single-end: run on R1 only fastqc --outdir "${OUT_DIR}" --threads 2 --quiet "${r1}" & fidone
# Wait for all background jobs to finishwaitecho "FastQC complete. Reports in ${OUT_DIR}"The & at the end of each FastQC call sends it to the background, so multiple samples run simultaneously. wait at the end blocks until all background jobs finish. On a machine with 16 cores, this will run 8 pairs of samples in parallel, with each FastQC job using 2 threads.
For HPC environments with SLURM, wrap the same logic in a job array:
#!/usr/bin/env bash#SBATCH --job-name=fastqc_array#SBATCH --time=00:30:00#SBATCH --cpus-per-task=2#SBATCH --mem=4G#SBATCH --array=1-30%10
# Load module on clustermodule load FastQC/0.12.1
FASTQ_LIST="sample_list.txt" # one file path per lineFASTQ_FILE=$(sed -n "${SLURM_ARRAY_TASK_ID}p" "${FASTQ_LIST}")OUT_DIR="results/fastqc"
mkdir -p "${OUT_DIR}"fastqc --outdir "${OUT_DIR}" --threads "${SLURM_CPUS_PER_TASK}" "${FASTQ_FILE}"The --array=1-30%10 directive submits 30 tasks but runs at most 10 concurrently, which is courteous to other users on shared clusters and stays within most queue limits.
Running MultiQC
Once FastQC has finished, running MultiQC is a single command. Point it at the directory containing your FastQC outputs and it finds the ZIP files automatically.
# Basic usage: scan results/fastqc and write report to results/multiqcmultiqc \ results/fastqc/ \ --outdir results/multiqc \ --filename multiqc_rnaseq_report \ --title "RNA-Seq QC: Experiment 2026-04" \ --forceThe --force flag overwrites any existing report with the same name. The --title flag embeds a label in the report header, which is useful when you generate multiple MultiQC reports across projects and need to tell them apart six months later.
MultiQC also aggregates outputs from tools beyond FastQC. If you have already run STAR, Salmon, or Trimmomatic, point MultiQC at the parent results directory and it will parse all of them automatically.
# Aggregate FastQC + STAR + Salmon in one reportmultiqc \ results/fastqc/ \ results/star/ \ results/salmon/ \ --outdir results/multiqc \ --filename multiqc_full_pipeline \ --force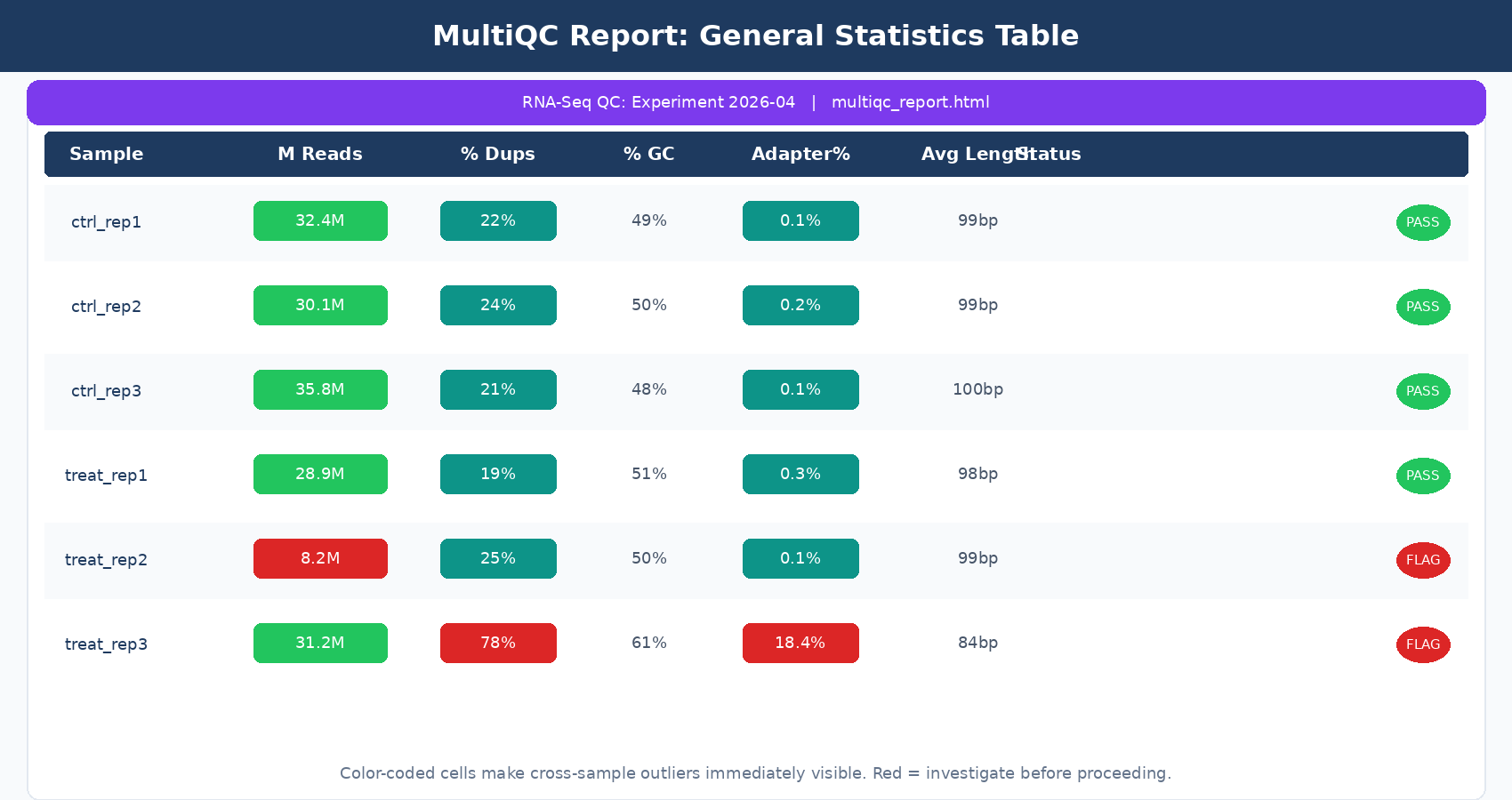
What to Look At in the MultiQC Report
Opening the MultiQC HTML report, the General Statistics table at the top is your first stop. Every row is a sample; every column is a metric pulled from whichever tools you included. For a standard FastQC-only run, the columns you care about are:
% Dups: duplication rate. In RNA-seq this is expected to be higher than in WGS, especially for poly-A selected libraries with highly expressed genes. A rate above 60 to 70% across all samples is worth noting, but is not automatically a failure.
% GC: GC percentage. Samples from the same species and library prep should cluster tightly here. An outlier sample with a dramatically different GC content is a contamination flag.
M Seqs: total read count in millions. For human RNA-seq, you want at least 20 million reads per sample for a standard differential expression study. Samples below 10 million will have poor sensitivity for lowly expressed genes.
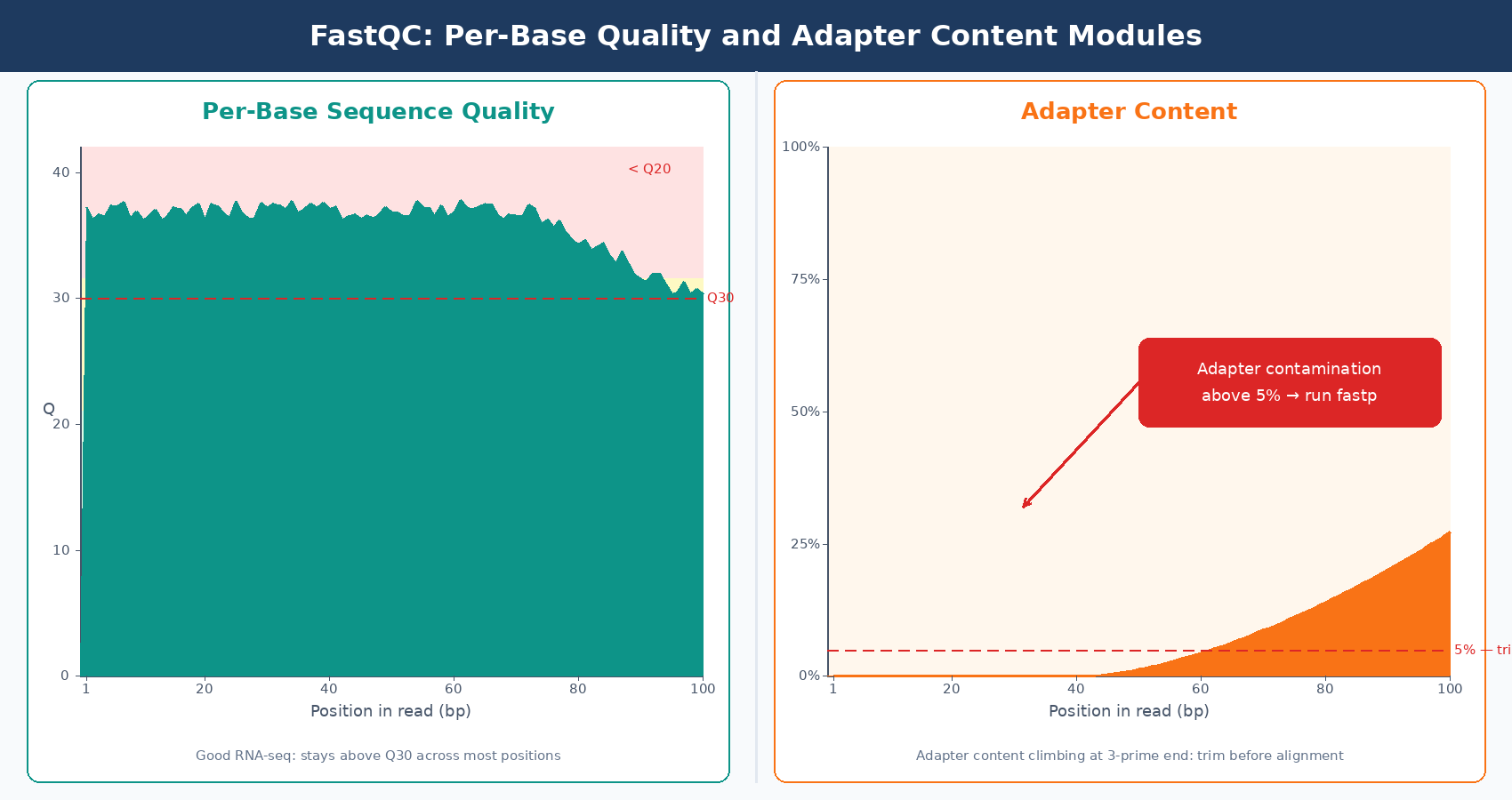
Scrolling through the module-specific sections, the per-base quality plot and the adapter content section are the two that actually drive decisions about trimming. If adapter content is near zero across all samples, you can skip trimming. If it climbs above 5 to 10% in the 3-prime end, trim before alignment.
GC content outliers between samples point to contamination or a sample swap
If one sample has 55% GC content while all others sit at 49%, that is not normal biological variation. It suggests contamination with a different organism, a pipetting error, or a sample label swap during library prep. Investigate before proceeding.
Parsing MultiQC Output in Python
MultiQC’s machine-readable outputs are stored in the multiqc_data/ directory alongside the HTML report. The file multiqc_fastqc.txt is a tab-separated table with all FastQC metrics per sample. This is the file to parse when you want to programmatically flag failing samples or log QC metrics to a database.
import pandas as pd
# Load MultiQC's FastQC summary tablemqc_fastqc = pd.read_csv( "results/multiqc/multiqc_data/multiqc_fastqc.txt", sep="\t", index_col="Sample")
# Define thresholds for RNA-seq QCthresholds = { "total_sequences": 10_000_000, # minimum 10M reads "pct_gc": (40, 65), # acceptable GC range "pct_duplication": 70, # flag if above 70% "avg_sequence_length": 50, # flag if trimmed too aggressively}
# Identify samples that fail each thresholdfailing = {}
for sample, row in mqc_fastqc.iterrows(): issues = []
if row["total_sequences"] < thresholds["total_sequences"]: issues.append( f"Low read count: {row['total_sequences']:,.0f} reads" )
gc = row["%GC"] if not (thresholds["pct_gc"][0] <= gc <= thresholds["pct_gc"][1]): issues.append(f"GC content out of range: {gc:.1f}%")
if row["pct_duplication"] > thresholds["pct_duplication"]: issues.append( f"High duplication: {row['pct_duplication']:.1f}%" )
if issues: failing[sample] = issues
# Report flagged samplesif failing: print(f"\n{'='*50}") print(f" {len(failing)} sample(s) flagged for review") print(f"{'='*50}") for sample, issues in failing.items(): print(f"\n {sample}:") for issue in issues: print(f" - {issue}")else: print("All samples passed QC thresholds.")Running this after MultiQC gives you a plain-text flag report. In a production pipeline, you would write the flags to a CSV, send an email alert, or integrate with a LIMS system. The key insight is that multiqc_data/ contains structured, machine-readable files for everything MultiQC parsed, not just FastQC. After a full pipeline run, multiqc_star.txt contains alignment statistics and multiqc_salmon.txt contains quantification metrics, all parseable with the same pattern.
A Complete QC Automation Script
Here is a single Python script that ties the full QC workflow together: run FastQC in parallel, run MultiQC, then parse the outputs and produce a QC summary table. This is the kind of script you add to a project’s scripts/ directory and run once at the start of every new experiment.
#!/usr/bin/env python3"""qc_pipeline.py
Automated RNA-seq QC: FastQC + MultiQC + flag parsing.Usage: python qc_pipeline.py --fastq-dir data/raw --out-dir results/qc"""
import subprocessimport argparseimport pandas as pdfrom pathlib import Pathfrom concurrent.futures import ThreadPoolExecutor, as_completed
def run_fastqc(fastq_file: Path, outdir: Path, threads: int = 2) -> str: """Run FastQC on a single FASTQ file and return the sample name.""" cmd = [ "fastqc", "--outdir", str(outdir), "--threads", str(threads), "--quiet", str(fastq_file) ] subprocess.run(cmd, check=True) return fastq_file.stem
def run_multiqc(fastqc_dir: Path, outdir: Path, title: str) -> None: """Aggregate FastQC reports with MultiQC.""" cmd = [ "multiqc", str(fastqc_dir), "--outdir", str(outdir), "--filename", "multiqc_report", "--title", title, "--force", "--quiet" ] subprocess.run(cmd, check=True)
def load_and_flag_qc(multiqc_data_dir: Path) -> pd.DataFrame: """Load MultiQC FastQC table and add pass/fail flags.""" mqc_file = multiqc_data_dir / "multiqc_fastqc.txt" df = pd.read_csv(mqc_file, sep="\t", index_col="Sample")
# Add boolean flag columns df["flag_low_reads"] = df["total_sequences"] < 10_000_000 df["flag_gc_outlier"] = ~df["%GC"].between(40, 65) df["flag_high_dups"] = df["pct_duplication"] > 70 df["any_flag"] = ( df["flag_low_reads"] | df["flag_gc_outlier"] | df["flag_high_dups"] )
return df[["total_sequences", "%GC", "pct_duplication", "flag_low_reads", "flag_gc_outlier", "flag_high_dups", "any_flag"]]
def main(): parser = argparse.ArgumentParser(description="RNA-seq QC pipeline") parser.add_argument("--fastq-dir", required=True, type=Path) parser.add_argument("--out-dir", required=True, type=Path) parser.add_argument("--threads", default=4, type=int) parser.add_argument("--title", default="RNA-Seq QC Report") args = parser.parse_args()
fastqc_dir = args.out_dir / "fastqc" multiqc_dir = args.out_dir / "multiqc" fastqc_dir.mkdir(parents=True, exist_ok=True) multiqc_dir.mkdir(parents=True, exist_ok=True)
# Discover FASTQ files fastq_files = sorted(args.fastq_dir.glob("*.fastq.gz")) print(f"Found {len(fastq_files)} FASTQ file(s). Running FastQC...")
# Run FastQC in parallel with ThreadPoolExecutor(max_workers=args.threads) as executor: futures = { executor.submit(run_fastqc, fq, fastqc_dir): fq for fq in fastq_files } for future in as_completed(futures): sample = future.result() print(f" Finished: {sample}")
# Run MultiQC print("\nRunning MultiQC...") run_multiqc(fastqc_dir, multiqc_dir, args.title)
# Parse and flag results print("\nParsing QC metrics...") qc_table = load_and_flag_qc(multiqc_dir / "multiqc_data")
flagged = qc_table[qc_table["any_flag"]] print(f"\nQC complete. {len(flagged)} sample(s) flagged.\n") print(qc_table.to_string())
# Save summary summary_path = args.out_dir / "qc_summary.tsv" qc_table.to_csv(summary_path, sep="\t") print(f"\nSummary saved to {summary_path}")
if __name__ == "__main__": main()Run it like this:
python qc_pipeline.py \ --fastq-dir data/raw \ --out-dir results/qc \ --threads 8 \ --title "Project XYZ RNA-Seq QC 2026-04"It will produce a fastqc/ folder with per-sample HTML reports, a multiqc/ folder with the aggregated report, and a qc_summary.tsv with flags for each sample.

When to Stop and Investigate
A good QC pass is not about hitting arbitrary thresholds. It is about understanding whether your samples are behaving consistently with each other and with the biology you expect.
Samples that differ dramatically from their replicates in total read count, GC content, or duplication rate deserve investigation before you proceed. The investigation question is: was this a wet lab failure, a sequencing run anomaly, or something biological? You cannot answer that from bioinformatics alone. Talk to whoever prepared the libraries.
If all your samples have a mapping rate of 85 to 92% except one at 45%, do not just exclude it and move on. Find out why. A wrong strandedness setting, a wrong reference genome, or genuine sample contamination are all plausible explanations, and only one of them justifies removal.
NotchBio runs FastQC and MultiQC automatically as part of its pipeline and surfaces the QC report alongside your differential expression results. If you would rather not maintain QC scripts across projects, it handles the full workflow from FASTQ upload to a flagged QC summary, without you writing or debugging a single bash line.
Further reading
Read another related post
Trimming Adapters with Trimmomatic and fastp: A Side-by-Side Walkthrough
When adapter trimming helps, when it hurts, and how to run Trimmomatic and fastp on RNA-seq data with the parameter choices that actually matter.
Research GuideRaw Reads to Counts: The Bulk RNA-Seq Pipeline Explained
A practical breakdown of every computational step in bulk RNA-seq: from FASTQ quality control through trimming, alignment, and quantification to your final count matrix.
Research GuideBatch Effects Will Ruin Your RNA-Seq Results
Batch effects silently corrupt bulk RNA-seq data. Learn how to detect them, why they happen, and which correction methods actually work.