Trimming Adapters with Trimmomatic and fastp: A Side-by-Side Walkthrough
Most RNA-seq tutorials include a trimming step without explaining why. You run Trimmomatic or fastp, produce trimmed FASTQ files, align them, and move on. The problem is that in many modern workflows, that trimming step is doing essentially nothing useful, and in some cases it is actively making your data worse by discarding reads that a decent aligner would have handled correctly.
This post is a working walkthrough of both tools: how to install them, the commands that matter, and the parameters worth thinking about. But it starts with the question that most tutorials skip: should you trim at all? If you have not already inspected your raw reads, start with How to Run FastQC and MultiQC on Raw RNA-Seq Reads. If you want the full pipeline context around where trimming fits, read Raw Reads to Counts: The Bulk RNA-Seq Pipeline Explained.
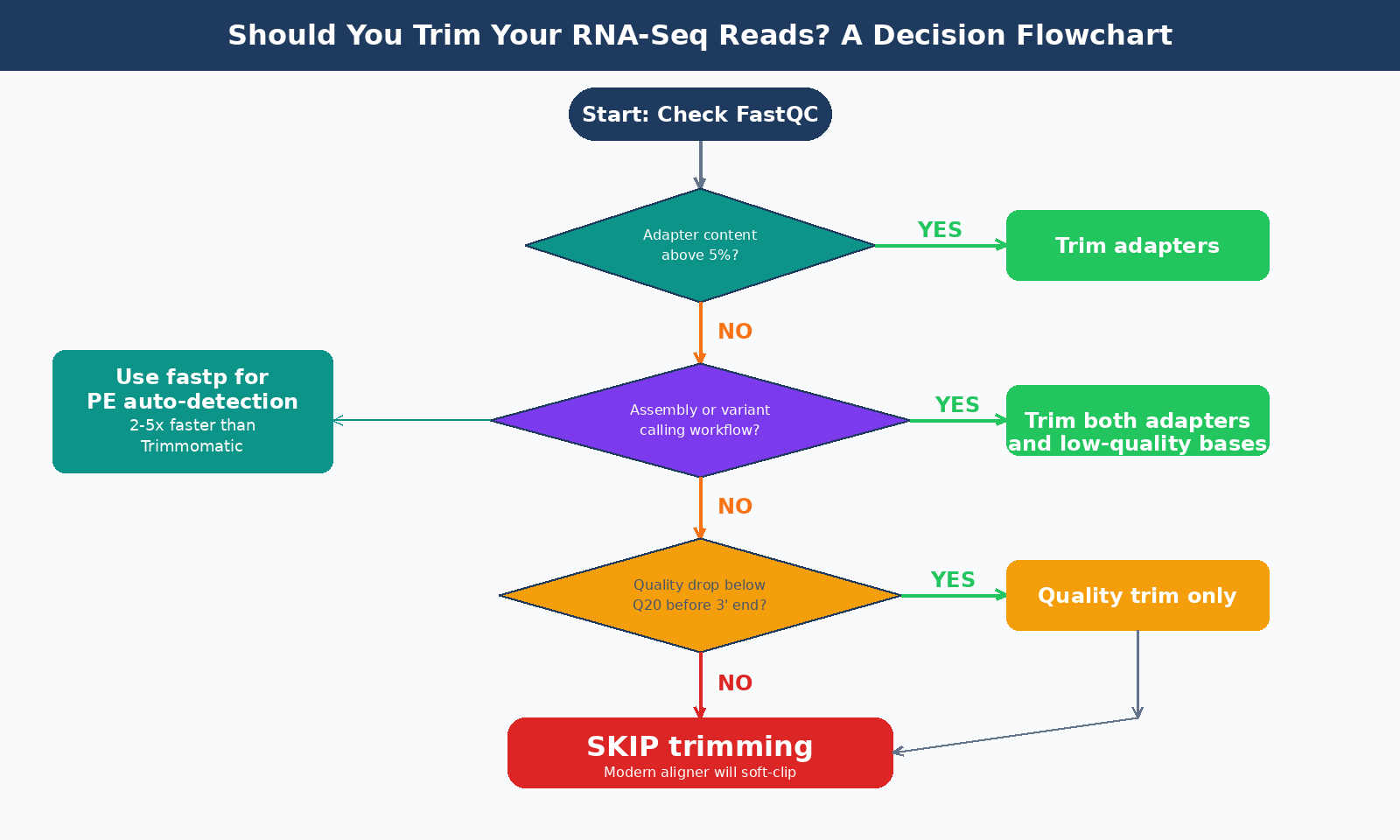
Should You Trim Your RNA-Seq Reads at All?
The honest answer is: it depends, and for many experiments the answer is no.
For counting applications such as differential gene expression analysis, read trimming is generally not required when using modern aligners. Tools like STAR, HISAT2, and the pseudo-aligner Salmon all handle adapter sequences correctly through soft-clipping: bases that cannot be mapped along with the majority of a read are marked and excluded from downstream analysis without needing to be removed beforehand. The adapter sequences are synthetic and do not exist in the reference genome, so a splice-aware aligner like STAR naturally soft-clips them out.
For RNA-seq, typical adapter contamination from a properly size-selected library falls between 0.2 and 2 percent of reads. At that level, the effect on alignment or quantification is negligible. Modern Illumina instruments and standard TruSeq library prep protocols rarely produce libraries with severe adapter contamination unless something went wrong in the size selection step.
Where trimming does matter:
Adapter content above 5 to 10 percent, confirmed in your FastQC adapter content module, is worth addressing. This typically happens with short insert libraries, miRNA data, or failed size selection.
Low-quality data from older runs, samples with degraded RNA, or runs on older instruments with poor quality at the 3-prime end can benefit from quality trimming even if adapter contamination is low.
Transcriptome assembly or variant calling workflows, where precise read ends matter, warrant trimming even on high-quality data.
Trimming is not a substitute for good library QC
If your FastQC report shows 30% adapter contamination, trimming will help your alignment. But you should also find out why a third of your reads contain adapter sequence. Size selection failure, very short inserts, or a degraded library are all possibilities worth investigating before you bank results on that sample.
Trimmomatic: The Veteran
Trimmomatic has been around since 2014 and was one of the first tools that worked reliably for Illumina trimming. It is Java-based, verbose in its configuration, and well understood. Its palindrome mode for paired-end data, which aligns the forward and reverse reads together with their adapter sequences using global alignment, is particularly effective at detecting adapter contamination down to a few bases of overlap.
Its main weakness in 2026 is its age. Trimmomatic has not seen a major update in several years, and it lacks features that have become standard: automatic adapter detection, UMI handling, and integrated QC output. It also does not scale especially well with threads on single-end data.
Installing Trimmomatic
conda install -c bioconda trimmomatic# orsudo apt install trimmomaticConfirm with trimmomatic -version. On some systems it is installed as a JAR file called directly:
java -jar /path/to/trimmomatic-0.39.jarRunning Trimmomatic: Paired-End
The standard paired-end command for a TruSeq stranded RNA-seq library:
trimmomatic PE \ -threads 8 \ -phred33 \ data/raw/sample_ctrl1_R1.fastq.gz \ data/raw/sample_ctrl1_R2.fastq.gz \ results/trimmed/sample_ctrl1_R1.paired.fastq.gz \ results/trimmed/sample_ctrl1_R1.unpaired.fastq.gz \ results/trimmed/sample_ctrl1_R2.paired.fastq.gz \ results/trimmed/sample_ctrl1_R2.unpaired.fastq.gz \ ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:2:True \ SLIDINGWINDOW:4:15 \ MINLEN:35The paired output produces four files: paired and unpaired reads for R1 and R2. Only the paired files are used in downstream alignment. The unpaired files contain reads whose partner was discarded during trimming. Most pipelines discard these.
Trimmomatic Parameters That Actually Matter
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:2:True is the adapter removal step. The parameters are, in order: the adapter FASTA file, seed mismatch tolerance, palindrome clip threshold, simple clip threshold, minimum adapter length, and whether to keep both reads when a palindrome is found. The defaults here are sensible for most experiments. The adapter FASTA files ship with Trimmomatic and live in its adapters/ directory. Match the file to your library prep: TruSeq3-PE.fa for paired-end TruSeq, TruSeq3-SE.fa for single-end, NexteraPE-PE.fa for Nextera.
SLIDINGWINDOW:4:15 scans with a 4-base window and cuts when average quality drops below Q15. This is a gentle quality threshold. Some labs use Q20, but for RNA-seq data destined for STAR alignment, aggressive quality trimming can introduce length bias without improving downstream DE analysis. Start at Q15 and check what it does to your mapping rate.
MINLEN:35 discards reads shorter than 35 bp after trimming. For 100bp reads this is conservative. For 75bp reads, you might lower it to 25. The key is that very short reads are more likely to multi-map and add noise to your count matrix than they are to provide signal.

fastp: The Modern Default
fastp is 2 to 5 times faster than other FASTQ preprocessing tools such as Trimmomatic or Cutadapt, despite performing far more operations than similar tools. It is written in C++ with multithreading baked in from the start, not bolted on. More importantly, it automatically detects adapter sequences for paired-end data by analyzing overlapping read pairs, which means you do not need to know or specify the adapter sequences in advance.
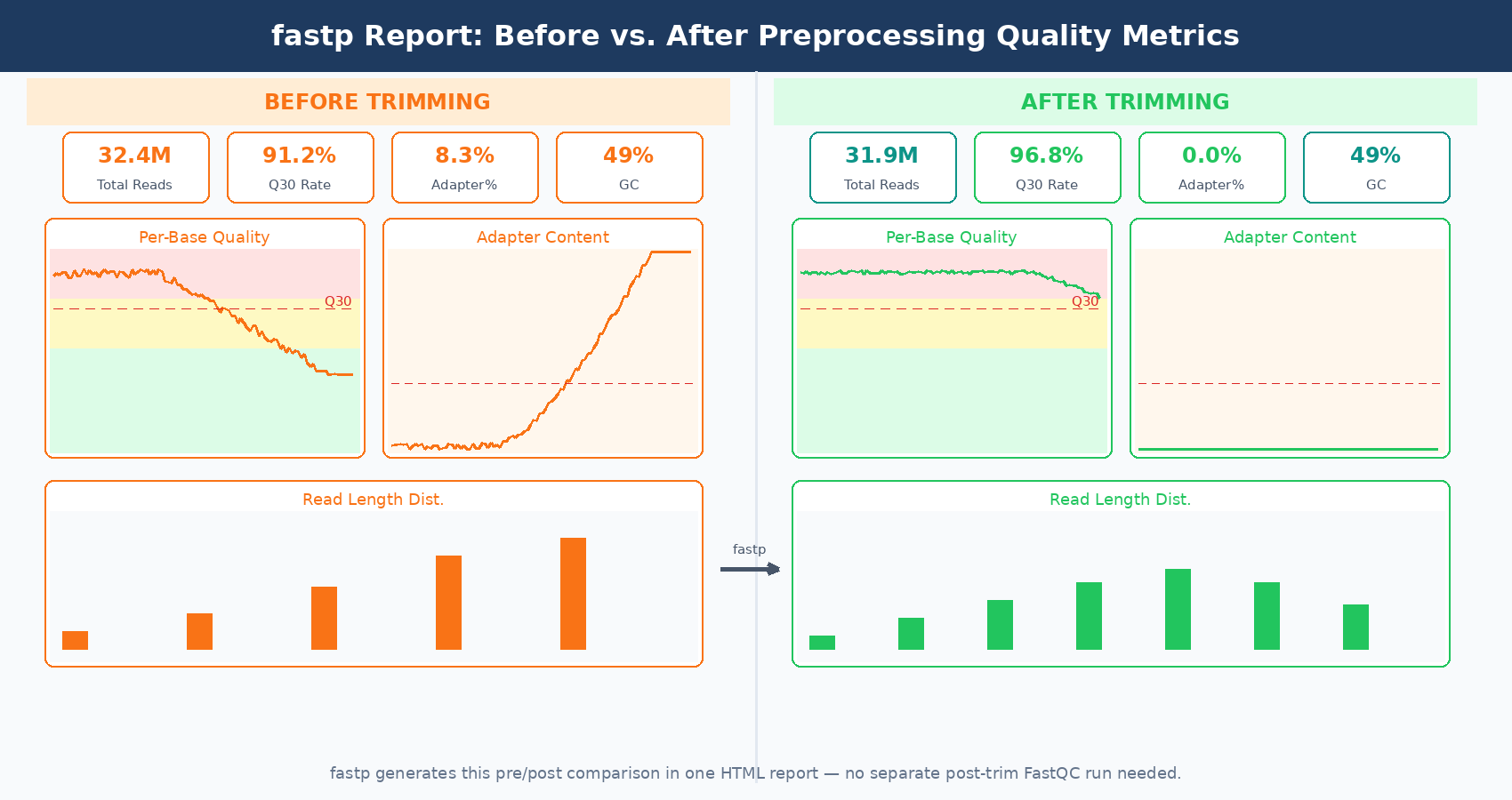
fastp also produces its own QC report for pre- and post-processed data in a single HTML page, giving you a before/after comparison without running a separate FastQC pass. For a lab running dozens of experiments a year, this reduces QC overhead noticeably.
Installing fastp
conda install -c bioconda fastpOr for the latest version directly:
wget http://opengene.org/fastp/fastpchmod a+x ./fastpCheck with fastp --version.
Running fastp: Paired-End
The equivalent command for a paired-end TruSeq RNA-seq library:
fastp \ --in1 data/raw/sample_ctrl1_R1.fastq.gz \ --in2 data/raw/sample_ctrl1_R2.fastq.gz \ --out1 results/trimmed/sample_ctrl1_R1.fastq.gz \ --out2 results/trimmed/sample_ctrl1_R2.fastq.gz \ --json results/qc/sample_ctrl1_fastp.json \ --html results/qc/sample_ctrl1_fastp.html \ --detect_adapter_for_pe \ --qualified_quality_phred 15 \ --length_required 35 \ --thread 8 \ --correctionFour key flags worth understanding here:
--detect_adapter_for_pe enables fastp’s overlap-based adapter detection for paired-end data. fastp finds the overlap between R1 and R2, then trims any bases that fall outside the overlapping region. This approach can trim adapters with as few as one base in the tail, outperforming sequence-matching tools in short-adapter scenarios.
--qualified_quality_phred 15 sets the per-base quality threshold. Bases below Q15 at the 3-prime end are trimmed. This mirrors the Trimmomatic SLIDINGWINDOW:4:15 behavior, though fastp’s algorithm is slightly different.
--length_required 35 is the minimum read length after trimming, equivalent to Trimmomatic’s MINLEN.
--correction enables base correction in overlapping read pairs. Where R1 and R2 disagree on a base in the overlapping region, fastp uses the higher-quality read to correct the lower-quality one. This is a small but real improvement in data quality that Trimmomatic does not offer.
fastp's --correction flag is safe to use for standard RNA-seq
Base correction in overlapping pairs is a genuine quality improvement with essentially no downside for standard differential expression workflows. The corrected bases are supported by the paired read at that position. Enable it unless you have a specific reason not to.
Running fastp: Single-End
For single-end libraries, adapter detection is less reliable without the paired read to anchor the overlap. It is worth specifying the adapter sequence explicitly if you know it:
fastp \ --in1 data/raw/sample_se.fastq.gz \ --out1 results/trimmed/sample_se.fastq.gz \ --adapter_sequence AGATCGGAAGAGCACACGTCTGAACTCCAGTCA \ --qualified_quality_phred 15 \ --length_required 25 \ --thread 8 \ --json results/qc/sample_se_fastp.json \ --html results/qc/sample_se_fastp.htmlThe adapter sequence above is the standard Illumina TruSeq forward read adapter. For Nextera libraries, replace it with CTGTCTCTTATACACATCT.
Automating Trimming Across Many Samples
For a multi-sample experiment, you want to run trimming in parallel rather than sequentially. Here is a bash script that trims all paired-end samples in a directory using fastp, then feeds the JSON outputs to MultiQC for a consolidated QC report:
#!/usr/bin/env bash# Usage: bash trim_all.sh data/raw results/trimmed results/qc
RAW_DIR="${1}"TRIM_DIR="${2:-results/trimmed}"QC_DIR="${3:-results/qc}"THREADS=8
mkdir -p "${TRIM_DIR}" "${QC_DIR}"
# Find R1 files and derive sample namesmapfile -t r1_files < <(find "${RAW_DIR}" -name "*_R1*.fastq.gz" | sort)
echo "Processing ${#r1_files[@]} sample(s) with fastp..."
for r1 in "${r1_files[@]}"; do r2="${r1/_R1/_R2}" # Extract sample name: remove directory path and suffix sample=$(basename "${r1}" | sed 's/_R1.*\.fastq\.gz//')
if [[ ! -f "${r2}" ]]; then echo " WARNING: R2 not found for ${sample}, skipping" continue fi
echo " Trimming: ${sample}"
fastp \ --in1 "${r1}" \ --in2 "${r2}" \ --out1 "${TRIM_DIR}/${sample}_R1.fastq.gz" \ --out2 "${TRIM_DIR}/${sample}_R2.fastq.gz" \ --json "${QC_DIR}/${sample}_fastp.json" \ --html "${QC_DIR}/${sample}_fastp.html" \ --detect_adapter_for_pe \ --qualified_quality_phred 15 \ --length_required 35 \ --thread "${THREADS}" \ --correction \ --disable_length_filtering \ 2>> "${QC_DIR}/${sample}_fastp.log" &done
waitecho "Trimming complete."
# Aggregate fastp JSON outputs into one MultiQC reportecho "Running MultiQC on fastp reports..."multiqc "${QC_DIR}" \ --outdir "${QC_DIR}/multiqc" \ --filename multiqc_trimming_report \ --force \ --quiet
echo "Done. MultiQC report at ${QC_DIR}/multiqc/multiqc_trimming_report.html"The & at the end of each fastp call parallelizes across samples. The wait blocks until all background jobs complete. MultiQC automatically recognizes fastp’s JSON output and incorporates the trimming statistics into the aggregated report, including pass/fail rates, adapter detection results, and before/after quality comparisons.
Side-by-Side: Trimmomatic vs. fastp
| Feature | Trimmomatic | fastp |
|---|---|---|
| Language | Java | C++ |
| Speed | Baseline | 2 to 5x faster |
| Auto adapter detection (PE) | No, requires FASTA file | Yes, overlap-based |
| Integrated QC report | No | Yes (HTML + JSON) |
| Base correction | No | Yes (--correction) |
| UMI handling | No | Yes |
| Active development | Stalled since 2020 | Actively maintained |
| MultiQC integration | Yes | Yes |
| Learning curve | Steeper | Gentler |
In practice, for a new RNA-seq project in 2026, fastp is the right default. It is faster, simpler to configure, produces its own QC output, and handles edge cases like short adapters better through its overlap-based detection. Trimmomatic remains a reasonable choice if you are maintaining an established pipeline, have a specific reason to prefer sequence-matching adapter removal, or are working in an environment where Java is already required for other tools.
One practical note: benchmarks show that Trimmomatic and AdapterRemoval, which use traditional sequence-matching, and fastp, which uses overlap analysis, all retain reads with the highest Q30 base percentages compared to other trimmers. Both approaches work. The difference is in speed, ease of use, and additional features.
Verify your results with a post-trimming FastQC check
Run FastQC on trimmed files the first time you use a new trimming configuration. Check that adapter content dropped to near zero and that your read length distribution looks as expected. If reads are being aggressively shortened across the board, your quality threshold may be too strict for the data. Adjust and recheck before scaling to all samples.
What to Check After Trimming
The two metrics that confirm trimming worked as expected are adapter content and read length distribution. In FastQC run on the trimmed files, adapter content should drop to near zero. Read length distribution should cluster tightly near your original read length if adapter contamination was low, or show a distribution shift toward shorter reads if significant trimming occurred.
A mapping rate check after alignment is the ultimate validation. If trimming improved your data quality, your STAR alignment rate should be equal to or slightly above the pre-trimming rate. If it dropped, something went wrong: you may have discarded too many reads or set your minimum length too aggressively. The before/after alignment rate comparison is a more meaningful quality metric than any of the intermediate trimming statistics.
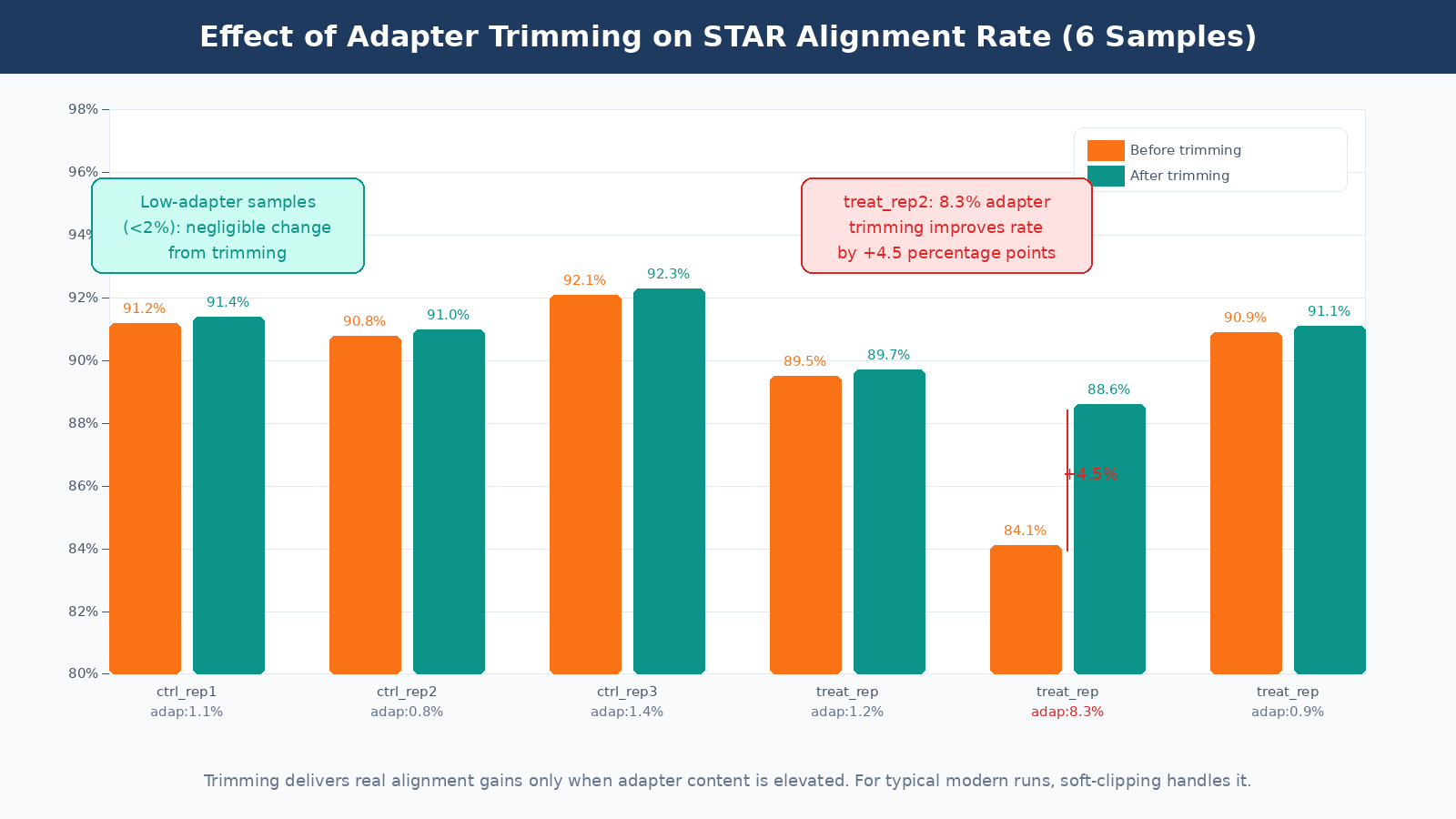
The Bottom Line
Check your FastQC adapter content before deciding to trim. For most modern Illumina RNA-seq runs with TruSeq libraries and proper size selection, adapter contamination will be low and trimming will have minimal effect on alignment or DE results. Modern aligners like STAR handle low-level adapter content through soft-clipping without any preprocessing.
When trimming is warranted, fastp is the current practical default. It is faster, self-documenting through its JSON output, and requires less configuration than Trimmomatic. For paired-end data, its overlap-based adapter detection works without specifying adapter sequences explicitly, which removes one class of configuration error entirely.
NotchBio runs fastp as part of its preprocessing pipeline with sensible default parameters, and surfaces the pre- and post-trimming QC metrics alongside the alignment statistics in the pipeline report. If you want to see what trimming does to your specific data without running and configuring the tools yourself, uploading your FASTQ files and reviewing the QC report is the fastest way to get that information.
Further reading
Read another related post
How to Run Differential Expression in Python with PyDESeq2
Complete PyDESeq2 tutorial: build a count matrix from Salmon output, fit a DeseqDataSet, run Wald tests, apply apeGLM shrinkage, and export DEG results in Python. No R required.
TutorialHow to Run DESeq2 in R: From Salmon Counts to DEG Results
Complete DESeq2 tutorial in R: import Salmon quant.sf files with tximeta, build a DESeqDataSet, run the Wald test, apply apeglm shrinkage, and export a ranked DEG table.
TutorialHow to Build a Counts Matrix from featureCounts and Salmon in Python
Python tutorial: parse featureCounts output, aggregate Salmon quant.sf files, build a tx2gene map from a GTF, round estimated counts, and save a DESeq2-ready integer count matrix with pandas.