Build a Counts Matrix from featureCounts and Salmon
Every RNA-seq pipeline eventually reaches the same bottleneck: you have counts, but they are spread across twelve separate files with columns you do not need, gene IDs in a format your downstream tool does not accept, and fractional values that DESeq2 will refuse to model.
Building a clean count matrix is not glamorous work. But it is the step that most tutorials skip over, which is why so many DESeq2 runs fail with cryptic errors about non-integer inputs, mismatched sample names, or missing genes.
This tutorial covers both paths in Python. If you aligned with STAR, you will load featureCounts output. If you quantified with Salmon, you will aggregate the quant.sf files. Either way, the final output is an integer gene-by-sample count matrix saved as a CSV, ready for DESeq2 in R or PyDESeq2 in Python.
We use the rnaseq conda environment from the Ubuntu and macOS setup guide.
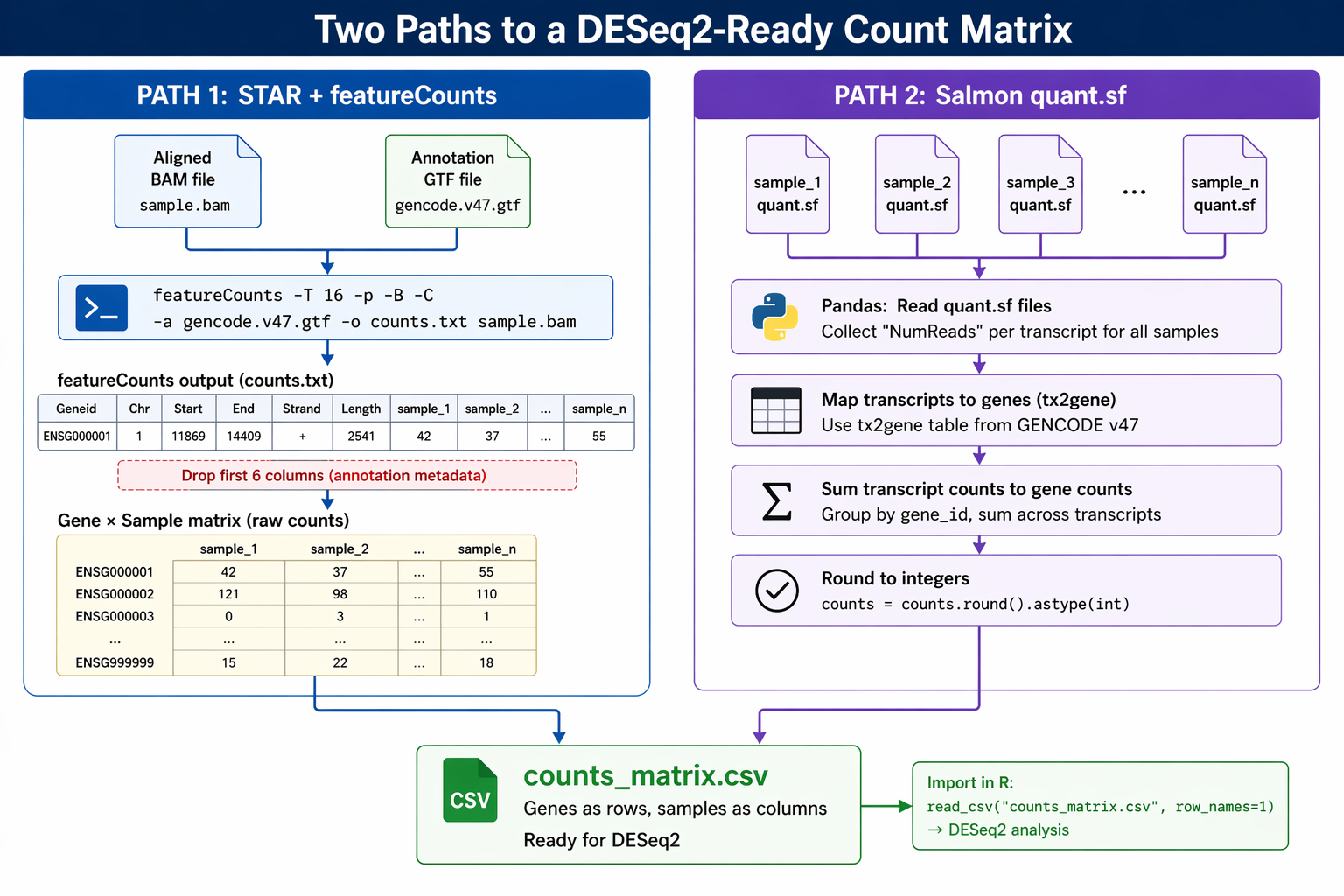
Path A: Loading featureCounts Output into a pandas DataFrame
featureCounts writes all samples into a single file. From the seventh column onward, the read count results for each sample are shown. Columns 1 through 6 contain gene identifier, chromosome, start, end, strand, and length information. You drop those first six columns and keep only the counts.
Run featureCounts on your BAM files
If you have not run featureCounts yet, this is the command. Run it from your project root after completing the STAR alignment.
conda activate rnaseq
GTF="references/gencode.v47.annotation.gtf"OUTFILE="results/counts/featurecounts_output.txt"
mkdir -p results/counts
# Run featureCounts on all STAR BAM filesfeatureCounts \ -p \ -t exon \ -g gene_id \ -a "$GTF" \ -o "$OUTFILE" \ -T 8 \ -s 2 \ results/star/*Aligned.sortedByCoord.out.bam
echo "featureCounts complete: $OUTFILE"The -p flag counts paired-end fragments instead of individual reads. -t exon summarizes at the exon level and aggregates to gene level. -s 2 specifies reverse-strand counting for standard Illumina TruSeq stranded libraries. Use -s 0 for unstranded libraries.
Set strandedness correctly or silently halve your counts
The -s parameter must match your library protocol. For unstranded libraries use -s 0. For TruSeq stranded libraries (dUTP method, most common) use -s 2. For first-strand protocols use -s 1. If you set this wrong, featureCounts will run without error but count reads from the wrong strand, typically losing around 50% of your reads. Check the .summary output file and confirm that “Assigned” exceeds 60%.
Load the featureCounts output in Python
import pandas as pd
# Load the featureCounts output file# The first line is a comment with the command; skip it with comment="#"fc_raw = pd.read_csv( "results/counts/featurecounts_output.txt", sep = "\t", comment = "#", # skip the command-line header comment index_col = 0 # gene ID becomes the index)
print(f"Raw featureCounts shape: {fc_raw.shape}")# (58780, 17) — 58780 genes, 6 annotation cols + 11 sample cols
print(fc_raw.columns.tolist()[:8])# ['Chr', 'Start', 'End', 'Strand', 'Length',# 'results/star/ctrl_1_Aligned.sortedByCoord.out.bam',# 'results/star/ctrl_2_Aligned.sortedByCoord.out.bam', ...]
# Drop the annotation columns (Chr, Start, End, Strand, Length)counts_fc = fc_raw.drop(columns=["Chr", "Start", "End", "Strand", "Length"])
# Clean up sample names: strip the full path and the BAM suffixcounts_fc.columns = ( counts_fc.columns .str.replace(r"results/star/", "", regex=False) .str.replace(r"_Aligned\.sortedByCoord\.out\.bam", "", regex=True))
print(f"Cleaned counts shape: {counts_fc.shape}")print(counts_fc.head(3))# ctrl_1 ctrl_2 ctrl_3 treat_1 treat_2 treat_3# ENSG00000000003.14 42 38 51 87 91 79# ENSG00000000419.12 523 498 541 612 634 601# ENSG00000000457.13 289 271 304 198 187 201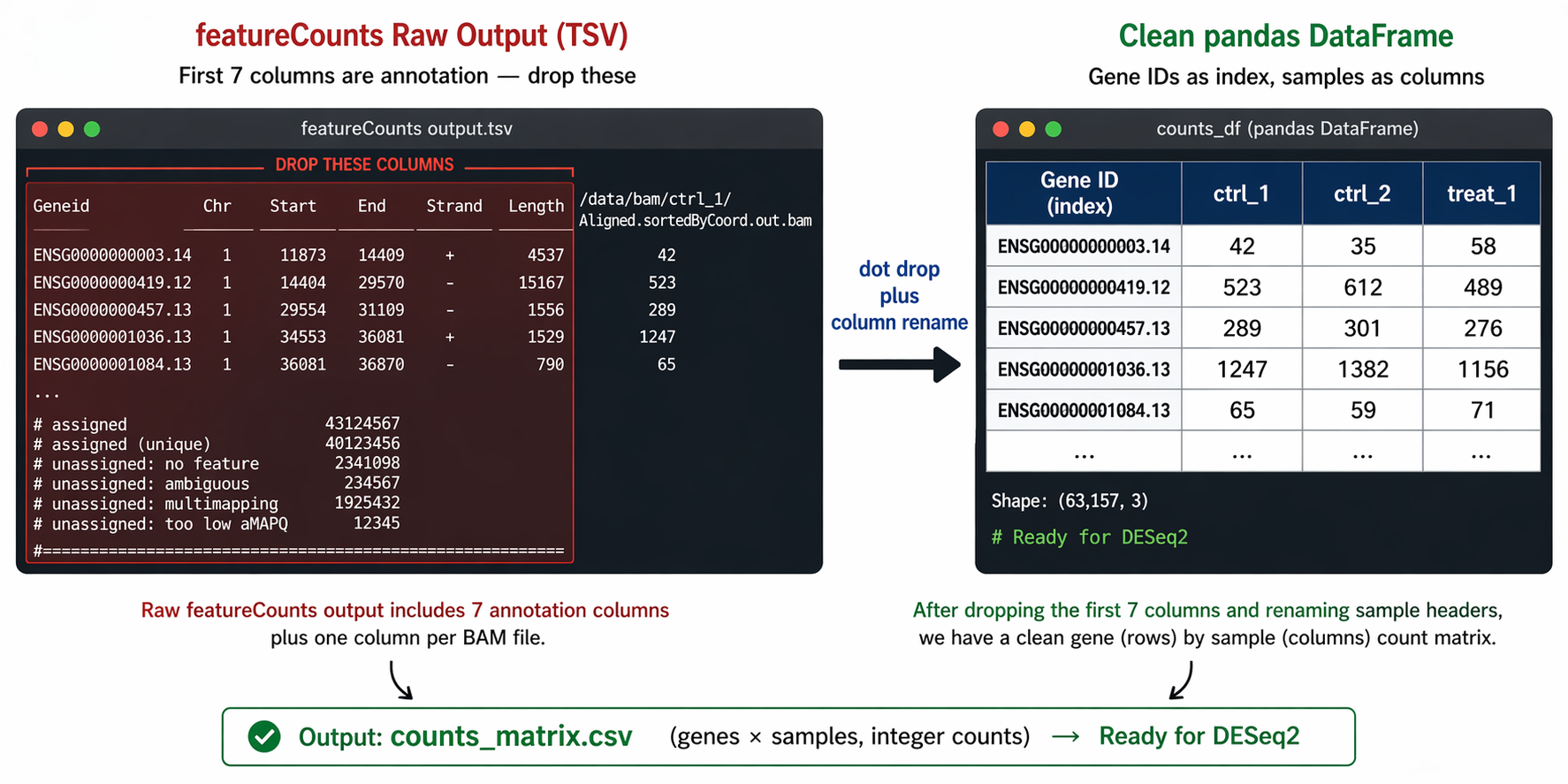
Path B: Aggregating Salmon quant.sf Files into a Count Matrix
Salmon produces one quant.sf file per sample. Each file has one row per transcript, not per gene. You need to load all files, aggregate from transcript to gene level using a tx2gene map, and round the estimated counts to integers.
Step 1: Load all quant.sf files
from pathlib import Path
# Find all quant.sf filesquant_dirs = sorted(Path("results/quant").glob("*_quant"))sample_names = [d.name.replace("_quant", "") for d in quant_dirs]
print(f"Found {len(sample_names)} samples: {sample_names[:4]}")
# Load each quant.sf and extract the NumReads columnparts = []for sample, qdir in zip(sample_names, quant_dirs): qf = pd.read_csv( qdir / "quant.sf", sep = "\t", index_col = "Name", usecols = ["Name", "NumReads"] ).rename(columns={"NumReads": sample}) parts.append(qf)
# Merge into a single transcript-level count matrixtx_counts = pd.concat(parts, axis=1)
print(f"Transcript-level matrix: {tx_counts.shape}")# (251638, 12) for GENCODE v47 with 12 samplesprint(tx_counts.head(3))Step 2: Build a transcript-to-gene map from the GENCODE GTF
def build_tx2gene(gtf_path: str) -> pd.DataFrame: """ Parse a GENCODE GTF and return a DataFrame mapping transcript_id to gene_id and gene_name. """ rows = [] with open(gtf_path) as fh: for line in fh: if line.startswith("#") or "\ttranscript\t" not in line: continue attrs = line.strip().split("\t")[8]
# Parse key "value" pairs from the attribute field attr_dict = {} for item in attrs.split(";"): item = item.strip() if " " in item: key, val = item.split(" ", 1) attr_dict[key] = val.strip('"')
tx = attr_dict.get("transcript_id", "") gene = attr_dict.get("gene_id", "") name = attr_dict.get("gene_name", "")
if tx and gene: rows.append({"transcript_id": tx, "gene_id": gene, "gene_name": name})
return pd.DataFrame(rows).drop_duplicates(subset="transcript_id")
tx2gene = build_tx2gene("references/gencode.v47.annotation.gtf")print(f"tx2gene rows: {len(tx2gene)}")# 246,624 unique transcripts
print(tx2gene.head(3))# transcript_id gene_id gene_name# ENST00000456328.2 ENSG00000223972.5 DDX11L2# ENST00000450305.2 ENSG00000223972.5 DDX11L2# ENST00000488147.1 ENSG00000227232.5 WASH7PCache the tx2gene table to avoid re-parsing the GTF every run
The GTF parse above takes 30 to 90 seconds for GENCODE v47. Save the result to a CSV and load it on subsequent runs: tx2gene.to_csv("references/tx2gene_v47.csv", index=False). Then replace the build_tx2gene() call with pd.read_csv("references/tx2gene_v47.csv").
Step 3: Aggregate transcript counts to gene level
# Salmon's GENCODE headers include pipe-separated fields: strip to transcript ID onlytx_counts.index = tx_counts.index.str.split("|").str[0]
# Map each transcript to its genetx2gene_dict = dict(zip(tx2gene["transcript_id"], tx2gene["gene_id"]))tx_counts["gene_id"] = tx_counts.index.map(tx2gene_dict)
# Report unmapped transcriptsn_unmapped = tx_counts["gene_id"].isna().sum()print(f"Unmapped transcripts: {n_unmapped} of {len(tx_counts)}")# Unmapped transcripts: 423 of 251638 (novel/unannotated transcripts)
# Drop unmapped and sum to gene levelgene_counts_salmon = ( tx_counts.dropna(subset=["gene_id"]) .groupby("gene_id") .sum(numeric_only=True))
print(f"Gene-level Salmon counts: {gene_counts_salmon.shape}")# (62703, 12)Rounding, Validating, and Saving the Count Matrix
Salmon outputs fractional estimated counts, not integers. DESeq2 requires integer counts. Round with .round().astype(int).
This step applies to the Salmon path only. featureCounts already produces integers.
# Salmon path: round estimated counts to integerscounts_salmon = gene_counts_salmon.round().astype(int)
# Verify no negative values crept in (should never happen, but check)assert (counts_salmon >= 0).all().all(), "Negative counts found"
# Check for all-zero genes (genes with no counts across any sample)n_zero_genes = (counts_salmon.sum(axis=1) == 0).sum()print(f"All-zero genes: {n_zero_genes}")
# Both paths: run these checksdef validate_count_matrix(counts: pd.DataFrame, source: str) -> None: """Quick sanity checks before saving.""" print(f"\n=== Validating {source} counts ===") print(f"Shape: {counts.shape}") print(f"Dtype: {counts.dtypes.unique()}") print(f"Min value: {counts.min().min()}") print(f"Max value: {counts.max().max()}") print(f"NaN cells: {counts.isna().sum().sum()}") print(f"All-zero genes: {(counts.sum(axis=1) == 0).sum()}") print(f"Median depth: {counts.sum(axis=0).median():.0f}")
validate_count_matrix(counts_fc, "featureCounts")validate_count_matrix(counts_salmon, "Salmon")Save the count matrix
import os
os.makedirs("results/counts", exist_ok=True)
# featureCounts pathcounts_fc.to_csv("results/counts/featurecounts_matrix.csv")
# Salmon pathcounts_salmon.to_csv("results/counts/salmon_gene_matrix.csv")
print("Count matrices saved.")print(" results/counts/featurecounts_matrix.csv")print(" results/counts/salmon_gene_matrix.csv")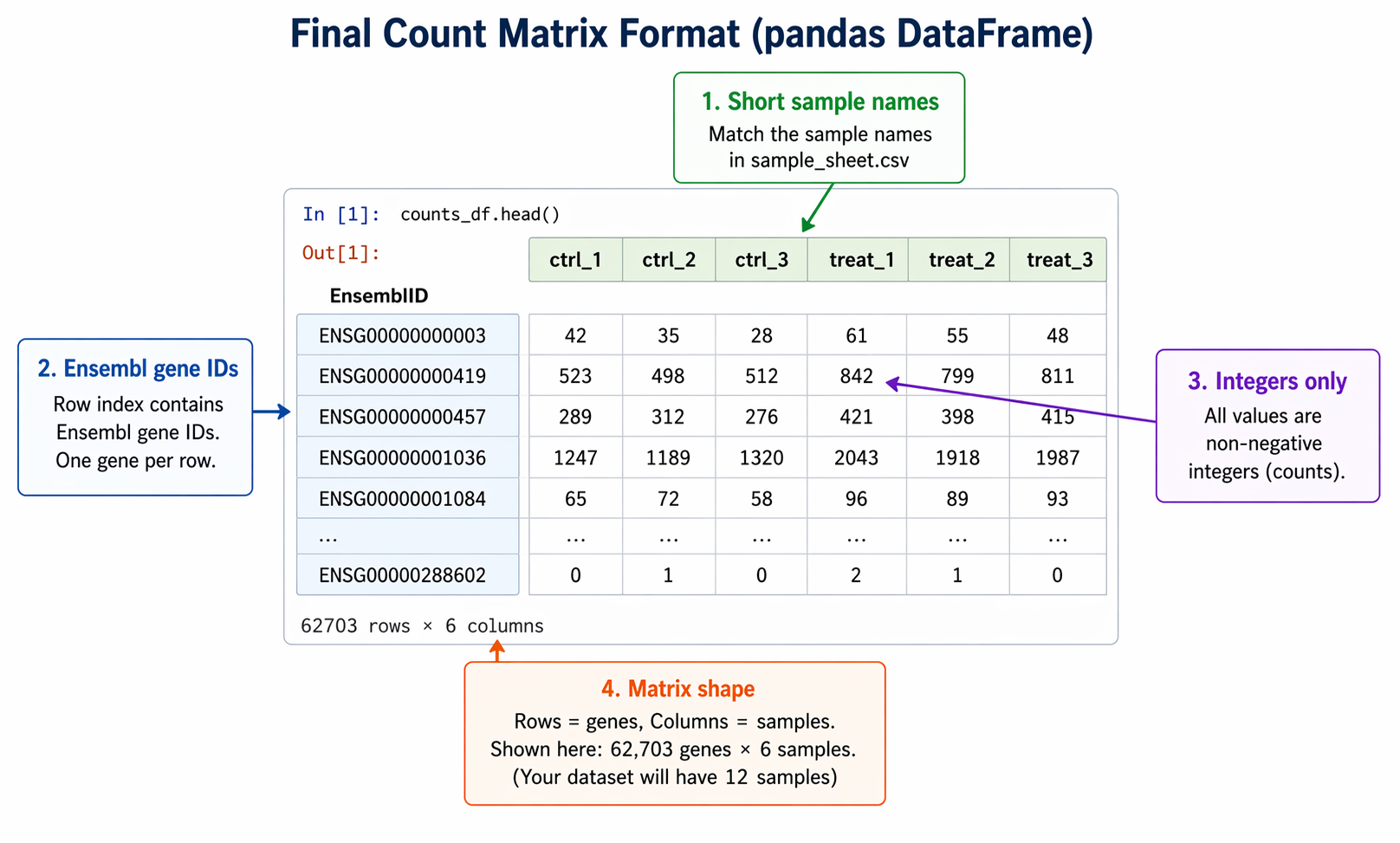
Filtering Low-Count Genes Before DESeq2
You do not have to filter in Python. DESeq2 handles filtering internally. But removing genes that have essentially no expression across all samples speeds up computation and reduces the multiple testing burden.
A standard filter: keep genes with at least 10 counts in at least 3 samples.
def filter_low_counts(counts: pd.DataFrame, min_count: int = 10, min_samples: int = 3) -> pd.DataFrame: """ Keep genes with at least min_count in at least min_samples samples. The min_samples value should match the size of your smallest group. """ keep = (counts >= min_count).sum(axis=1) >= min_samples filtered = counts.loc[keep] print(f"Genes before filter: {len(counts)}") print(f"Genes after filter: {len(filtered)}") return filtered
# Apply to either matrix before downstream analysiscounts_fc_filtered = filter_low_counts(counts_fc)counts_salmon_filtered = filter_low_counts(counts_salmon)
# Save filtered versionscounts_fc_filtered.to_csv("results/counts/featurecounts_filtered.csv")counts_salmon_filtered.to_csv("results/counts/salmon_filtered.csv")Aligning with Your Sample Sheet
The column order in your count matrix must match the row order of your sample metadata. A mismatch here is silent in DESeq2 but catastrophic for your results.
# Load sample metadatametadata = pd.read_csv("sample_sheet.csv", index_col="sample_id")print("Sample sheet order:")print(metadata.index.tolist())
# Reorder count matrix columns to match the sample sheetcounts_final = counts_salmon_filtered.loc[:, metadata.index]
# Confirm alignmentassert list(counts_final.columns) == list(metadata.index), \ "Sample order mismatch between count matrix and metadata"
print("Sample alignment verified.")print(f"Final matrix: {counts_final.shape}")# Final matrix: (19847, 12)From here, the matrix is ready for DESeq2. In R, load it with read.csv() and pass it to DESeqDataSetFromMatrix(). In Python, load it with pd.read_csv() and transpose before passing to DeseqDataSet(). Both approaches are covered in How to Run DESeq2 in R and From Salmon Output to DEGs in Python with PyDESeq2.
Manual Count Matrix Assembly vs NotchBio
The steps above are genuinely tedious: featureCounts flag choices, GTF parsing, GENCODE header stripping, rounding, validation, and sample alignment. Getting any one of them wrong produces a count matrix that looks correct but gives you wrong differential expression results.
NotchBio assembles the count matrix automatically. Whether you quantified with Salmon or aligned with STAR, the platform converts the outputs into a validated integer matrix, checks sample alignment against your metadata, and passes it directly to the differential expression pipeline.
| Step | Manual Python | NotchBio |
|---|---|---|
| Run featureCounts | featureCounts -p -t exon -g gene_id -s 2 | Automatic after STAR alignment |
| Drop annotation columns | drop(columns=["Chr","Start","End","Strand","Length"]) | Not required |
| Clean BAM path from column names | str.replace() regex | Not required |
| Load Salmon quant.sf files | Loop over 12 dirs, pd.concat() | Automatic |
| Parse GTF for tx2gene | Custom parser function, 30-90 sec | Pre-built for GENCODE v47, Ensembl |
| Strip GENCODE pipe headers | `str.split(" | ").str[0]` |
| Aggregate transcript to gene | groupby("gene_id").sum() | Automatic |
| Round estimated counts | .round().astype(int) | Automatic |
| Validate matrix | Manual assertion loop | Built-in, errors surface before DE runs |
| Align with sample sheet | reindex(metadata.index) with assertion | Auto-verified from upload metadata |
| Time to clean count matrix | 30-60 min for 12 samples | 2 min after quantification finishes |
If you want to skip the pandas plumbing and go directly to differential expression, notchbio.app handles count matrix assembly as part of every analysis. Upload your Salmon outputs or STAR alignments, and the validated matrix is ready before you finish writing your methods section.
Further reading
Read another related post
The One-Bioinformatician Problem: Stop Being The Bottleneck
If you are the only bioinformatician serving multiple PIs, you are the bottleneck. Here is how to scale with templates, self-service, and clear handoffs.
TutorialORA vs GSEA: A Side-by-Side Tutorial in R with clusterProfiler
ORA and GSEA answer different questions. A working clusterProfiler tutorial with FDR correction, proper backgrounds, and side-by-side result interpretation.
Research GuideWhy Your DESeq2 Log2 Fold Change Cutoff Of Zero Is Wrong
Filtering DEGs at log2FC greater than zero returns half your genome. How to choose a defensible cutoff, apply lfcShrink, and avoid the GO-term explosion.