Why Your DESeq2 Log2 Fold Change Cutoff Of Zero Is Wrong
A researcher posted to the bioinformatics community with a specific problem: too many GO terms. Their differential expression analysis had returned hundreds of significant gene ontology hits, and they could not figure out what to report. The top comment identified the issue immediately. They had filtered their DESeq2 results with abs(log2FoldChange) > 0. That cutoff does not filter anything. Any gene with a non-zero fold change passes it, which in a real experiment is nearly every gene that was tested. The GO-term explosion was not a downstream analysis problem. It was a upstream filtering problem disguised as an enrichment problem.
This mistake is common enough to have its own community thread. Understanding why it happens, and how to replace it with something defensible, is one of the most practically useful things you can do for the quality of your published results.
Why log2FC Greater Than Zero Means Nothing
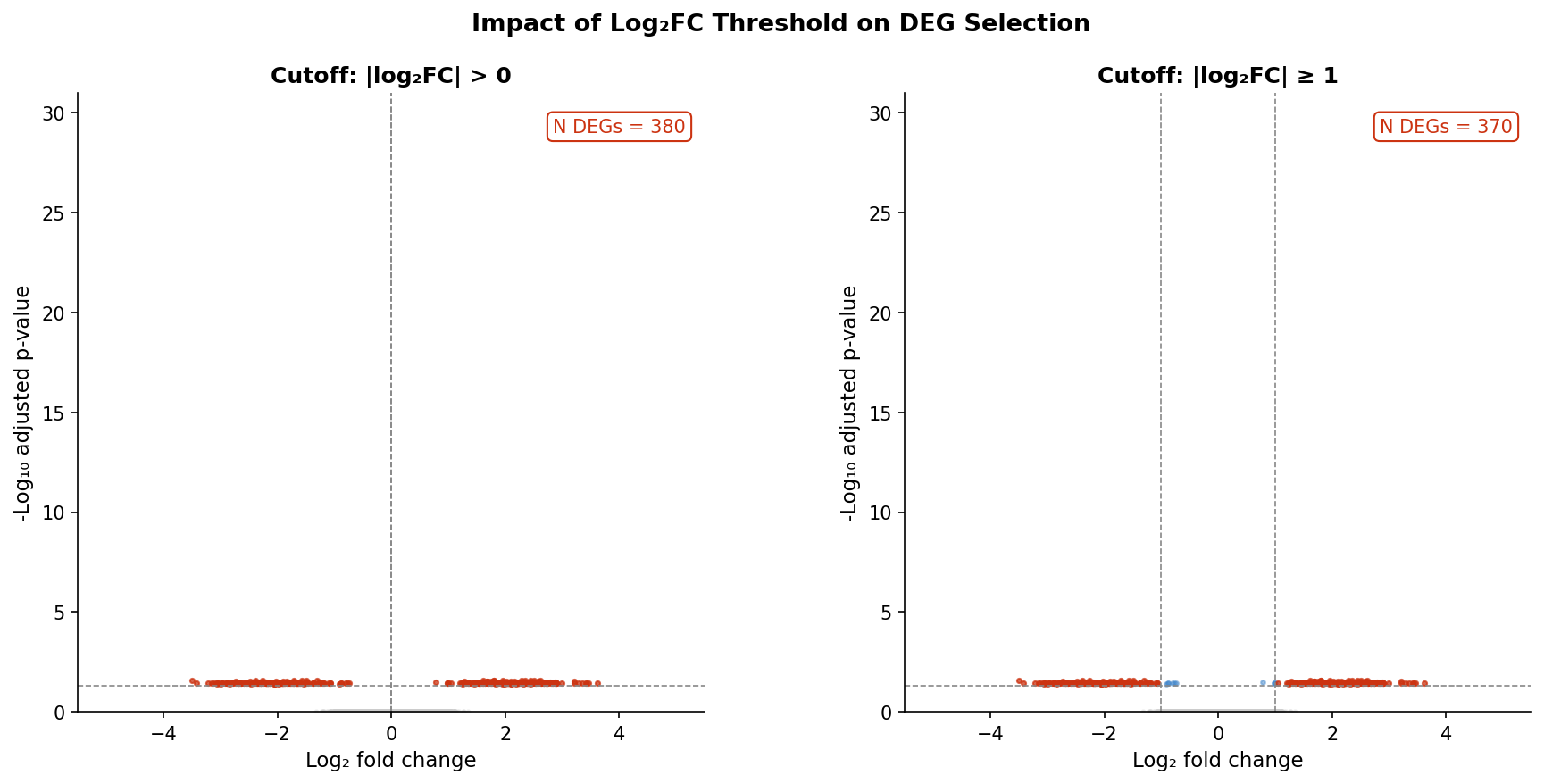
Fold change on a log2 scale is symmetric around zero. A log2FC of 1 means a twofold upregulation. A log2FC of minus 1 means a twofold downregulation. A log2FC of 0.01 means a 0.7 percent increase. It is statistically distinguishable from zero, but it is biologically indistinguishable from noise in almost any RNA-seq experiment.
When you filter abs(log2FoldChange) > 0, you are asking DESeq2 to return every gene whose estimated fold change is not exactly zero. In practice, because DESeq2 estimates fold changes from count data with biological variability, essentially no gene returns exactly zero. The filter is tautological. You are left with whatever the padj threshold alone selects, which at padj < 0.05 in a well-powered experiment can be thousands of genes.
Those thousands of genes then go into your enrichment analysis. The enrichment analysis tests every GO term in the database against a gene list that is far too broad to be biologically informative. Terms that are enriched simply because your gene list is large enough to contain a representative fraction of every annotation class come out as significant. The results are not wrong in a statistical sense; they are just not telling you anything about your experiment specifically.
What Reviewers Expect and Why
The question a reviewer asks when they see your fold change cutoff is: what is the smallest biological effect you are willing to call meaningful? A cutoff of zero says your answer is “any effect at all.” That is not a scientific position. It is an absence of one.
The cutoffs that are defensible to reviewers are in the range of abs(log2FoldChange) >= 1 (twofold change) or abs(log2FoldChange) >= 1.5 (roughly threefold). A twofold threshold has a long history in gene expression analysis predating RNA-seq, which gives it a kind of conventional legitimacy. More importantly, it corresponds to an effect size that has a reasonable chance of biological consequence. Not every twofold change matters biologically, but almost nothing below twofold is worth discussing in most contexts.
For some experiments, particularly those using very precise perturbations where you expect subtle transcriptional changes, a lower cutoff with a strong justification is defensible. The key word is justification. You need to be able to explain why you chose the threshold you chose, and “because it was the default” is not a justification.
The combination that works for most bulk RNA-seq papers is padj < 0.05 and abs(log2FoldChange) >= 1. Both conditions must be met. The adjusted p-value guards against false positives. The fold change guards against statistical but biologically trivial effects.
The defensible defaults for DEG filtering
Filter on both thresholds simultaneously: adjusted p-value below 0.05 (never raw p-value) and absolute log2 fold change at or above 1. Both conditions must be satisfied for a gene to be reported as differentially expressed. If your experiment involves subtle expected effects and you need a lower fold change cutoff, state that explicitly in your methods and be prepared to justify it to reviewers.
padj Versus p: Never Filter on the Raw p-Value
The adjusted p-value and the raw p-value are both present in the DESeq2 results object. They look similar. They are not interchangeable.
The raw p-value from DESeq2 is the probability of observing a test statistic as extreme as the one measured, assuming no real effect, for that one gene. Across the tens of thousands of genes tested simultaneously, many raw p-values below 0.05 will appear purely by chance. This is the multiple testing problem.
The adjusted p-value corrects for the number of simultaneous tests using the Benjamini-Hochberg procedure. It controls the false discovery rate: if you filter at padj < 0.05, you expect at most 5 percent of the genes that pass to be false positives. That is the guarantee you need to make a claim about your gene list.
If you filter on the raw p-value column instead, you have no such guarantee. In a standard human RNA-seq experiment testing 20,000 genes at p < 0.05, you expect roughly 1,000 false positives by chance alone. Those will be in your DEG list and your enrichment analysis.
DESeq2 applies Benjamini-Hochberg correction by default. The column in the results table is padj. Filter on that column. The column named pvalue is for diagnostics and reporting, not for thresholding.
lfcShrink and Why It Changes Your Threshold Logic
Fold change estimation in DESeq2 without shrinkage is noisy for genes with low counts. A gene with 3 counts in one condition and 8 in another can show an apparent log2FC of 1.4, but the confidence interval on that estimate spans several log2FC units. The point estimate is unreliable.
lfcShrink addresses this by pulling fold change estimates for noisy, lowly expressed genes toward zero. The statistical significance of those genes (their padj) is unchanged. What changes is the reported fold change: it becomes a shrunken estimate that better reflects the precision of the underlying data.
This matters for your threshold logic in a specific way. If you are working with unshrunken fold changes, a threshold of abs(log2FoldChange) >= 1 will include some genes where the true fold change is much smaller and the observed value reflects sampling noise. After lfcShrink, the same threshold is more conservative: genes that pass it are more likely to have a genuine fold change near 1.
The practical recommendation is to apply lfcShrink using the apeglm estimator before filtering on fold change. The apeglm estimator is the current default recommendation from the DESeq2 team and performs well across a range of experimental designs.
library(DESeq2)library(apeglm)
# Run DESeq2dds <- DESeq(dds)
# Apply shrinkage to the contrast of interest# "condition_treated_vs_control" must match the resultsNames(dds) outputres_shrunk <- lfcShrink(dds, coef = "condition_treated_vs_control", type = "apeglm")
# Filter using the shrunken fold changes and adjusted p-valuedeg_table <- as.data.frame(res_shrunk)degs <- deg_table[ !is.na(deg_table$padj) & deg_table$padj < 0.05 & abs(deg_table$log2FoldChange) >= 1, ]
nrow(degs) # Number of DEGs after both thresholds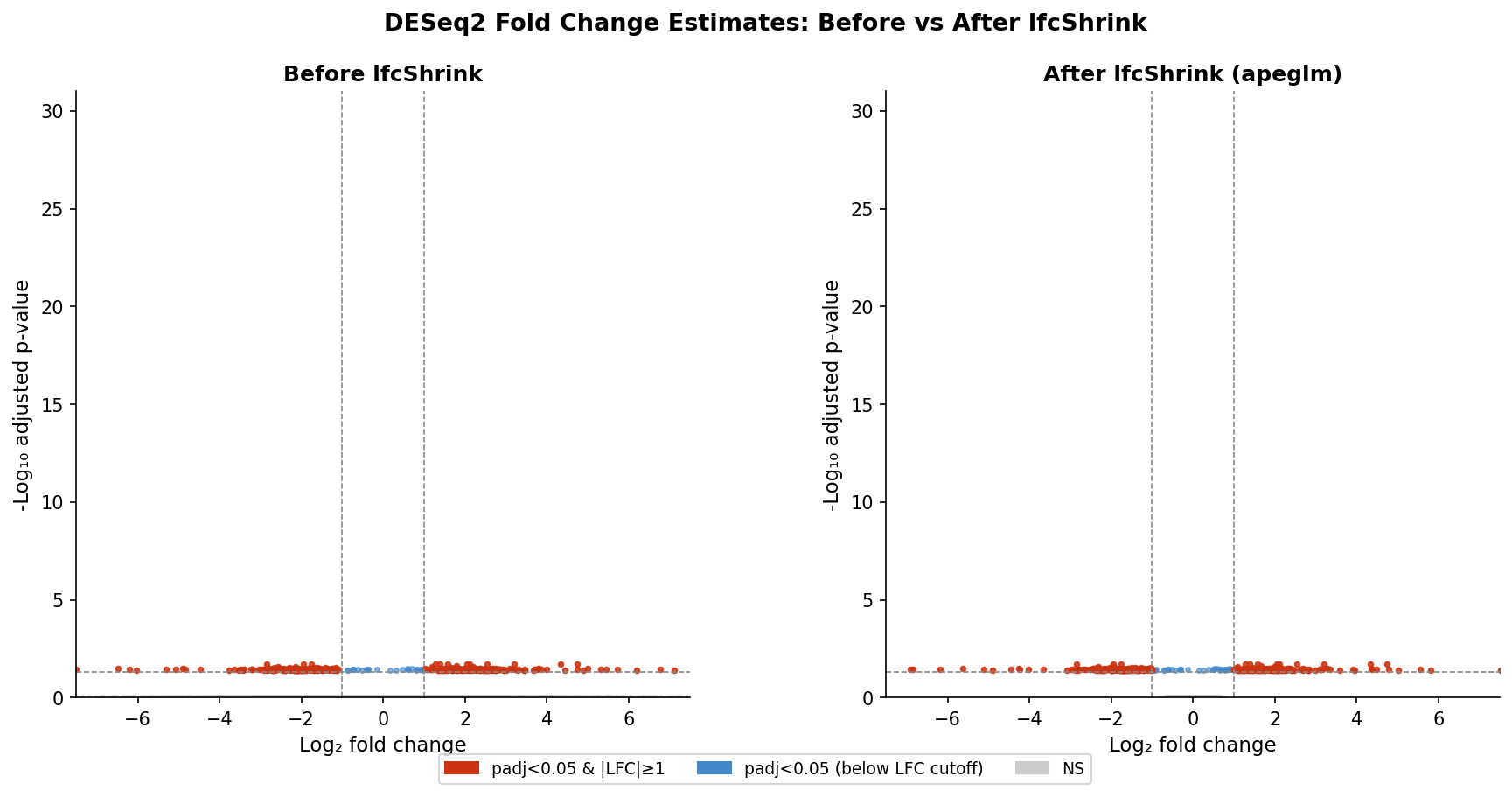
Statistical Significance Versus Biological Significance
The most important conceptual distinction in differential expression analysis is between statistical significance and biological significance. They measure different things. Confusing them produces both false positives and false negatives in the interpretation of results.
A gene is statistically significant when its padj is below your chosen threshold. That means: given the data you collected, the probability that this effect is a false positive is acceptably low. It says nothing about whether the effect is large enough to matter biologically.
A gene is biologically significant when the magnitude of the change is large enough to have a plausible functional consequence. Whether a twofold change in a given gene is biologically meaningful depends entirely on the gene and the context. For a transcription factor, a twofold increase in expression might drive major downstream regulatory changes. For a structural protein, a twofold change might have no measurable phenotype.
The fold change threshold you apply at the DEG filtering step is a proxy for biological significance. It is a crude proxy; you should not claim that every gene above your threshold is biologically important or that every gene below it is irrelevant. What you can claim is that you have applied a principled filter that limits your reported gene list to effects large enough to warrant further investigation.
The right framing in a paper is: “Genes meeting both the statistical criterion (padj < 0.05) and the effect size criterion (abs(log2FoldChange) >= 1) were considered differentially expressed for downstream analysis.” That framing is accurate about what you did and defensible to a reviewer who asks why.
Putting It Together
The corrected workflow is three steps. Apply lfcShrink with the apeglm estimator to get reliable fold change estimates. Filter the results table requiring both padj < 0.05 and abs(log2FoldChange) >= 1. Pass that filtered gene list, and only that filtered gene list, to your enrichment analysis, with the full tested universe (all genes with non-NA padj) as the background.
Those three steps will not make your biology more interesting. What they will do is ensure that the results you report reflect your experiment rather than statistical artifacts of a misconfigured filter. They will also cut your GO-term list from hundreds of terms to a manageable set that a reader can actually interpret.
NotchBio applies these defaults automatically: lfcShrink with apeglm runs on every differential expression analysis, the significance filter requires both padj < 0.05 and abs(log2FoldChange) >= 1 by default, and the enrichment background is always the tested gene universe. If you want to change those defaults, you can. But the starting point is correct, which means you do not have to know to set it.
Related Reading
Further reading
Read another related post
Why Cell Line RNA-Seq Experiments Fail
Passage drift, undetected mycoplasma, serum lot changes, and pseudoreplication silently corrupt cell line RNA-seq. What each looks like and how to prevent it.
Research GuideSTAR vs HISAT2 vs Salmon: Which Aligner Should You Use?
STAR aligns to the genome, HISAT2 uses less memory, Salmon skips alignment. What each approach means for your RNA-seq results and when each is the right call.
Research GuideWhat Is GSEA and Why Does It Beat a Simple DEG List
How GSEA finds coordinated pathway signals a DEG list misses: how the algorithm works, what NES and the leading edge mean, and how to run it with fgsea in R.