From Salmon Output to DEGs in Python with PyDESeq2
Most RNA-seq tutorials assume you are comfortable in R. If your lab runs everything in Python, the jump to R for a single DESeq2 step has always been an annoying cost.
PyDESeq2 fixes that. It is a from-scratch Python implementation of the DESeq2 pipeline, maintained by the Owkin team, that produces results numerically equivalent to the R version. Same median-of-ratios normalization, same dispersion trend fitting, same Wald tests, same apeGLM shrinkage.
This tutorial is the pipeline-focused walkthrough: the part most guides skip is getting from raw Salmon output to a clean count matrix, so that is where we spend our time. We load Salmon quant.sf files directly into a count matrix, build a DeseqDataSet, run the model, apply lfc_shrink(), and export a ranked DEG table. We continue from How to Build a Salmon Index and Quantify Bulk RNA-Seq Reads using your results/quant/ directory and the rnaseq conda environment from How to Set Up a Bulk RNA-Seq Analysis Environment on Ubuntu and macOS.
If you want the full PyDESeq2 feature reference rather than the Salmon-to-DEG path — including multi-factor designs and running several contrasts from one model — see Differential Expression Analysis in Python with PyDESeq2: A Complete Tutorial. And if you specifically want to confirm the Python results match R DESeq2, PyDESeq2 vs R DESeq2: Validation and the scanpy Workflow walks through a side-by-side comparison.

Why PyDESeq2 Exists and When to Use It
R DESeq2 is still the reference implementation. It has been cited over 60,000 times and is the most battle-tested differential expression tool in the field. If you already have an R pipeline that works, there is no reason to rewrite it in Python.
PyDESeq2 matters for three situations.
First, labs with a pure Python stack. If your upstream processing, visualization, and downstream analysis all happen in Python, PyDESeq2 removes the friction of switching languages for one step.
Second, integration with Python-native tools. PyDESeq2 plays well with scanpy, anndata, and the broader single-cell ecosystem. A DeseqDataSet is an AnnData subclass, so the same object can be passed to scanpy utilities.
Third, teams that want reproducibility without the R dependency burden. Bioconductor packages can be brittle to install, especially on Apple Silicon. A pure pip install pydeseq2 avoids that entire maintenance surface.
What PyDESeq2 does (and does not) replicate
PyDESeq2 implements the core DESeq2 pipeline: size factors, dispersion estimation, Wald tests, Cook’s filtering, independent filtering, apeGLM shrinkage, and multifactor designs. It does not currently implement the likelihood ratio test, rlog transformation, or some of the more niche R-side features. For the vast majority of two-group or multifactor differential expression analyses, it is a drop-in replacement.
How to Install PyDESeq2 in a Clean conda Environment
PyDESeq2 is a pip-only package. Install it into your rnaseq conda environment from the Ubuntu and macOS setup guide, or create a dedicated environment.
# Dedicated environment (recommended for first-time users)conda create -n pydeseq2 python=3.11 -yconda activate pydeseq2
# Install PyDESeq2 and the plotting stackpip install pydeseq2==0.5.4pip install pandas numpy matplotlib seaborn mygeneVerify the install:
import pydeseq2print(pydeseq2.__version__)How to Build a Count Matrix from Salmon quant.sf in Python
PyDESeq2 takes a raw counts matrix as input. You need to convert Salmon’s per-sample quant.sf files into a single DataFrame.
There are two paths. The simple one uses pandas directly. The more rigorous one uses pyroe or tximeta equivalents for proper transcript-to-gene aggregation. For a standard bulk RNA-seq analysis, the pandas path is sufficient.
The pandas-native approach
import pandas as pdimport numpy as npfrom pathlib import Path
# List all quant directoriesquant_dirs = sorted(Path("results/quant").glob("*_quant"))sample_names = [d.name.replace("_quant", "") for d in quant_dirs]
# Load each quant.sf and extract NumReads columncounts_list = []for sample, qdir in zip(sample_names, quant_dirs): qf = qdir / "quant.sf" df = pd.read_csv(qf, sep="\t") # Use transcript ID as index; take NumReads as the count column df = df.set_index("Name")[["NumReads"]].rename(columns={"NumReads": sample}) counts_list.append(df)
# Merge all samples into one transcript-level matrixtranscript_counts = pd.concat(counts_list, axis=1)print(f"Transcript-level counts shape: {transcript_counts.shape}")# (251638, 12) for GENCODE v47 with 12 samplesAggregate to gene level using a tx2gene mapping
Salmon quantifies transcripts, not genes. DESeq2 models work at the gene level. Aggregate using a transcript-to-gene map built from the GENCODE GTF.
# Build tx2gene from the GENCODE GTF (downloaded when building the Salmon index)def build_tx2gene_from_gtf(gtf_path: str) -> pd.DataFrame: """Extract transcript_id -> gene_id mapping from a GENCODE GTF.""" rows = [] with open(gtf_path) as fh: for line in fh: if line.startswith("#") or "\ttranscript\t" not in line: continue attrs = line.strip().split("\t")[8] attr_dict = dict( item.strip().split(" ", 1) for item in attrs.split(";") if item.strip() ) tx = attr_dict.get("transcript_id", "").strip('"') gene = attr_dict.get("gene_id", "").strip('"') gene_name = attr_dict.get("gene_name", "").strip('"') if tx and gene: rows.append((tx, gene, gene_name)) return pd.DataFrame(rows, columns=["transcript_id", "gene_id", "symbol"])
tx2gene = build_tx2gene_from_gtf("references/gencode.v47.annotation.gtf")print(f"tx2gene mapping: {len(tx2gene)} transcripts")
# Note: Salmon uses the full GENCODE header as transcript ID (with pipes)# Strip the pipe-delimited extras to match the tx2gene keystranscript_counts.index = transcript_counts.index.str.split("|").str[0]Now aggregate transcript counts to gene level. We sum counts across all transcripts of a gene.
# Map transcripts to genestx_to_gene_dict = dict(zip(tx2gene["transcript_id"], tx2gene["gene_id"]))transcript_counts["gene_id"] = transcript_counts.index.map(tx_to_gene_dict)
# Drop transcripts that did not map (rare edge cases)transcript_counts = transcript_counts.dropna(subset=["gene_id"])
# Sum to gene levelgene_counts = transcript_counts.groupby("gene_id").sum()
# Round estimated counts to integers (DESeq2 requires integer counts)gene_counts = gene_counts.round().astype(int)
print(f"Gene-level counts shape: {gene_counts.shape}")# Expected ~62,703 genes for GENCODE v47 humanPyDESeq2 expects samples as rows, not columns
This is the biggest gotcha when moving from R to Python. R DESeq2 uses counts(dds) with genes as rows and samples as columns. PyDESeq2 reverses this. You must transpose your matrix so that rows are samples and columns are genes before passing it to DeseqDataSet.
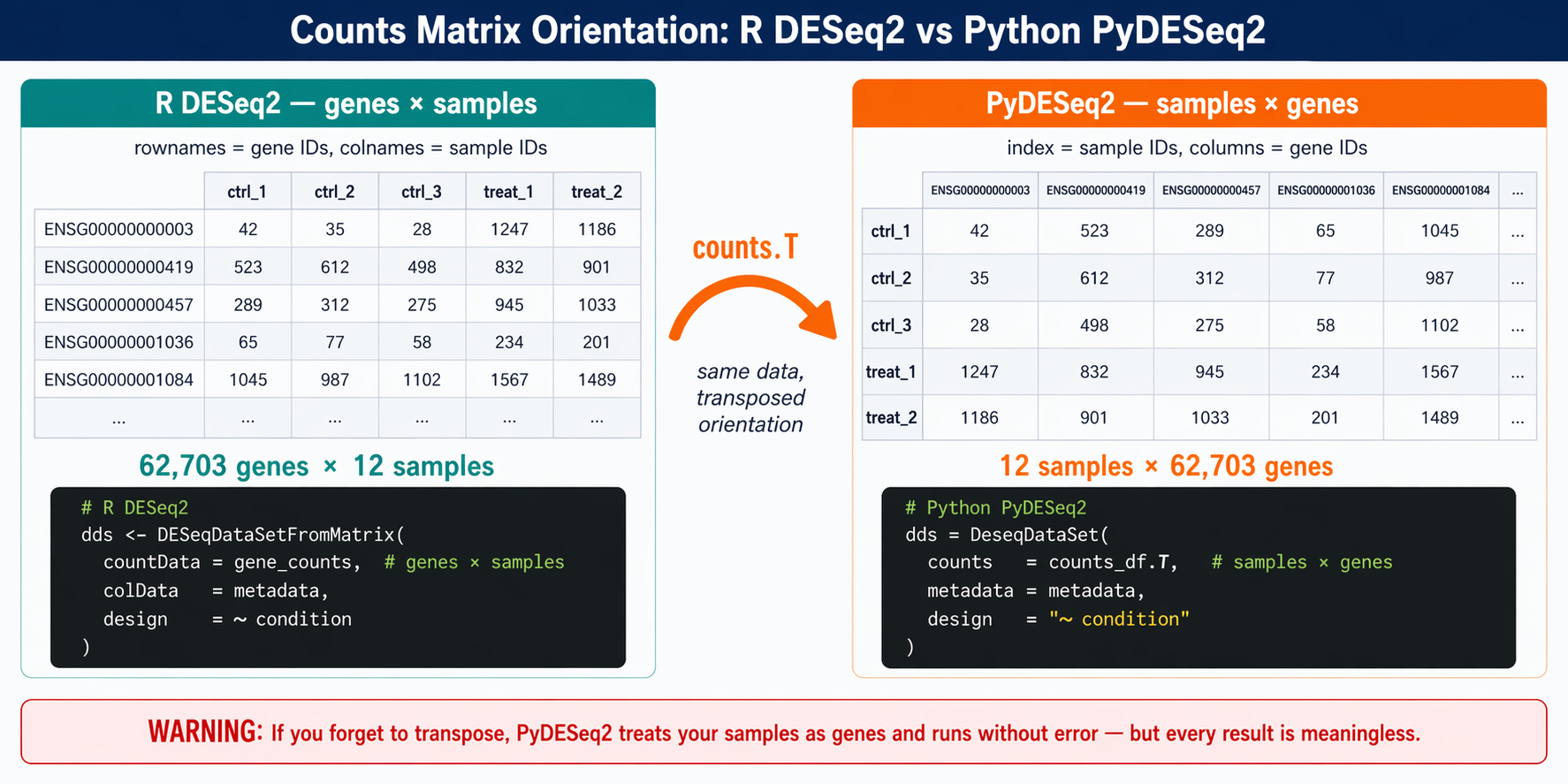
Transpose and align with metadata
# Transpose: samples × genes (PyDESeq2 convention)counts_df = gene_counts.Tcounts_df.index.name = "sample_id"
# Load sample metadata (from the GEO/SRA download step)metadata = pd.read_csv("sample_sheet.csv", index_col="sample_id")
# Ensure sample order matches between counts and metadatacounts_df = counts_df.loc[metadata.index]
# Sanity checkassert (counts_df.index == metadata.index).all(), "Sample order mismatch"print(f"Counts: {counts_df.shape} Metadata: {metadata.shape}")# Counts: (12, 62703) Metadata: (12, 3)How to Build a DeseqDataSet and Run deseq2()
With the counts matrix correctly oriented, building a DeseqDataSet is straightforward.
from pydeseq2.dds import DeseqDataSetfrom pydeseq2.default_inference import DefaultInference
# Filter out genes with very low counts (same logic as R)# Keep genes with at least 10 counts in at least 3 sampleskeep = (counts_df >= 10).sum(axis=0) >= 3counts_filtered = counts_df.loc[:, keep]print(f"Genes after filtering: {counts_filtered.shape[1]}")
# Configure inference (parallelism)inference = DefaultInference(n_cpus=8)
# Build the DeseqDataSetdds = DeseqDataSet( counts = counts_filtered, metadata = metadata, design = "~ condition", # formulaic syntax, same as R refit_cooks = True, inference = inference, quiet = False)
# Fit size factors, dispersion, and GLM coefficientsdds.deseq2()The deseq2() method prints progress as it runs.
Fitting size factors...... done in 0.2 seconds.
Fitting dispersions...... done in 4.3 seconds.
Fitting dispersion trend curve...... done in 0.1 seconds.
Fitting MAP dispersions...... done in 4.1 seconds.
Fitting LFCs...... done in 2.8 seconds.
Running Cook's outlier filtering...... done in 0.3 seconds.Each step corresponds exactly to the R DESeq2 pipeline. Size factor estimation uses median-of-ratios. Dispersion uses the same parametric trend fit with MAP shrinkage.

Multifactor designs
If MultiQC from the QC step or the PCA from a previous analysis showed a batch effect, add batch to the design. The formulaic syntax matches R.
# Multifactor design: control for batch, test conditiondds_multi = DeseqDataSet( counts = counts_filtered, metadata = metadata, design = "~ batch + condition", # batch is the covariate inference = inference,)dds_multi.deseq2()The last term in the formula is the tested variable. ~ batch + condition tests the condition effect after adjusting for batch.
How to Run Wald Tests with DeseqStats
The DeseqStats class runs the actual differential expression test. It takes a fitted DeseqDataSet and a contrast specification.
from pydeseq2.ds import DeseqStats
# Contrast format: [variable, tested_level, reference_level]# This computes LFC of treated vs controlds = DeseqStats( dds, contrast = ["condition", "treated", "control"], alpha = 0.05, inference = inference,)
# Run the full statistical analysisds.summary()The summary() method prints summary statistics and populates ds.results_df with the per-gene results.
Running Wald tests...... done in 1.8 seconds.
Log2 fold change & Wald test p-value: condition treated vs control baseMean log2FoldChange lfcSE stat pvalue padjENSG00000000003 42.31 0.108164 0.172823 0.625870 5.314148e-01 7.618301e-01ENSG00000000419 523.89 0.043257 0.096132 0.449980 6.527483e-01 8.340195e-01ENSG00000000457 289.74 -0.194851 0.152904 -1.274326 2.025813e-01 4.372941e-01...
out of 19,847 with nonzero total read countadjusted p-value < 0.05LFC > 0 (up) : 1,203, 6.1%LFC < 0 (down) : 954, 4.8%outliers : 18, 0.1%low counts : 2,087, 10.5%Access the results as a pandas DataFrame.
results = ds.results_dfprint(results.head())print(results.columns.tolist())# ['baseMean', 'log2FoldChange', 'lfcSE', 'stat', 'pvalue', 'padj']Contrast direction matters
The contrast ["condition", "treated", "control"] means LFC of treated vs control. Positive LFC = upregulated in treated. If you swap the order to ["condition", "control", "treated"], you get the same genes with flipped signs. Always document which direction is “up” in your results.
How to Apply apeGLM LFC Shrinkage
Raw maximum likelihood fold changes from DESeq2 are noisy for low-count genes. lfc_shrink() with apeGLM pulls these unreliable estimates toward zero while preserving well-supported large fold changes.
# Apply apeGLM shrinkage IN-PLACE on the ds objectds.lfc_shrink(coeff="condition_treated_vs_control")
# Confirm the LFCs were shrunkprint(f"LFCs shrunk: {ds.shrunk_LFCs}")# True
# The results_df is now updated with shrunken LFCsresults_shrunk = ds.results_dflfc_shrink() mutates results_df in place
Unlike R’s lfcShrink() which returns a new object, PyDESeq2’s lfc_shrink() overwrites ds.results_df with the shrunken values. If you want to compare before and after shrinkage, save the raw results first. Once shrunk, there is no way to recover the original fold changes without re-running the Wald tests.
The right pattern is to save a copy before shrinking.
# Save raw LFCs firstresults_raw = ds.results_df.copy()
# Then shrinkds.lfc_shrink(coeff="condition_treated_vs_control")results_shrunk = ds.results_df.copy()
# Now you can compareprint("Raw LFC range: ", results_raw["log2FoldChange"].min(), results_raw["log2FoldChange"].max())print("Shrunk LFC range:", results_shrunk["log2FoldChange"].min(), results_shrunk["log2FoldChange"].max())The coefficient name to pass to lfc_shrink
The coeff argument must match a column name in the internal LFC matrix. PyDESeq2 formats these as {variable}_{level}_vs_{reference}.
# List the available coefficientsprint(dds.LFC.columns.tolist())# ['Intercept', 'condition_treated_vs_control']For a simple two-group design, there will be one non-intercept coefficient. For multifactor designs, you will see multiple and need to pick the one matching your contrast.
How to Filter and Export Results with pandas
With results_shrunk in hand, filtering and exporting is standard pandas.
# Annotate with gene symbols using mygeneimport mygene
mg = mygene.MyGeneInfo()
# Strip version numbers from Ensembl IDsresults_shrunk["gene_id_clean"] = results_shrunk.index.str.split(".").str[0]
# Query in batch (1000 genes at a time for speed)gene_info = mg.querymany( results_shrunk["gene_id_clean"].tolist(), scopes = "ensembl.gene", fields = "symbol", species = "human", as_dataframe = True)symbol_map = gene_info["symbol"].to_dict()results_shrunk["symbol"] = results_shrunk["gene_id_clean"].map(symbol_map)
# Reorder columnsresults_shrunk = results_shrunk[[ "symbol", "baseMean", "log2FoldChange", "lfcSE", "stat", "pvalue", "padj"]]
# Sort by padj, tiebreaker by absolute LFCresults_shrunk = results_shrunk.sort_values( ["padj", "log2FoldChange"], ascending=[True, False], key=lambda c: c.abs() if c.name == "log2FoldChange" else c)Export full and filtered tables
from pathlib import PathPath("results/pydeseq2").mkdir(parents=True, exist_ok=True)
# Full resultsresults_shrunk.to_csv("results/pydeseq2/all_genes.csv")
# Significant DEGs: padj < 0.05 and |LFC| > 1degs = results_shrunk.query("padj < 0.05 and abs(log2FoldChange) > 1")degs.to_csv("results/pydeseq2/significant_degs.csv")
print(f"Total genes tested: {len(results_shrunk)}")print(f"Significant DEGs: {len(degs)}")print(f" Upregulated: {(degs['log2FoldChange'] > 0).sum()}")print(f" Downregulated: {(degs['log2FoldChange'] < 0).sum()}")A Complete Runnable Script
Save this as run_pydeseq2.py and run it end to end.
#!/usr/bin/env python3"""run_pydeseq2.py: Full PyDESeq2 pipeline from Salmon quant.sf to DEG table."""import pandas as pdfrom pathlib import Pathfrom pydeseq2.dds import DeseqDataSetfrom pydeseq2.ds import DeseqStatsfrom pydeseq2.default_inference import DefaultInference
# ── 1. Load counts from Salmon ───────────────────────────────────────quant_dirs = sorted(Path("results/quant").glob("*_quant"))samples = [d.name.replace("_quant", "") for d in quant_dirs]
parts = []for s, q in zip(samples, quant_dirs): df = pd.read_csv(q / "quant.sf", sep="\t") df.index = df["Name"].str.split("|").str[0] parts.append(df[["NumReads"]].rename(columns={"NumReads": s}))tx_counts = pd.concat(parts, axis=1)
# ── 2. Aggregate to gene level ───────────────────────────────────────tx2gene = pd.read_csv("references/tx2gene.csv") # precomputedtx_counts["gene_id"] = tx_counts.index.map( dict(zip(tx2gene.transcript_id, tx2gene.gene_id)))gene_counts = ( tx_counts.dropna(subset=["gene_id"]) .groupby("gene_id").sum() .round().astype(int))
# ── 3. Transpose + metadata ──────────────────────────────────────────counts_df = gene_counts.Tmetadata = pd.read_csv("sample_sheet.csv", index_col="sample_id")counts_df = counts_df.loc[metadata.index]
# Filter low-count geneskeep = (counts_df >= 10).sum(axis=0) >= 3counts_df = counts_df.loc[:, keep]
# ── 4. Fit DeseqDataSet ──────────────────────────────────────────────inference = DefaultInference(n_cpus=8)dds = DeseqDataSet( counts=counts_df, metadata=metadata, design="~ condition", inference=inference, quiet=False)dds.deseq2()
# ── 5. Wald test + shrinkage ─────────────────────────────────────────ds = DeseqStats(dds, contrast=["condition", "treated", "control"], inference=inference)ds.summary()ds.lfc_shrink(coeff="condition_treated_vs_control")
# ── 6. Export ────────────────────────────────────────────────────────out = Path("results/pydeseq2"); out.mkdir(parents=True, exist_ok=True)ds.results_df.to_csv(out / "all_genes.csv")ds.results_df.query("padj < 0.05 and abs(log2FoldChange) > 1") \ .to_csv(out / "significant_degs.csv")
print("Done. Results in results/pydeseq2/")Run it:
conda activate pydeseq2python run_pydeseq2.pyManual PyDESeq2 vs NotchBio
The PyDESeq2 pipeline above is clean and fully reproducible in Python. It still requires managing a conda environment, writing the count matrix aggregation code, and handling the samples × genes orientation correctly.
NotchBio runs the equivalent analysis automatically. Upload your Salmon outputs or let the platform run Salmon for you, then select the comparison you want. The counts matrix construction, model fitting, shrinkage, and annotation all happen server-side.
| Step | Manual PyDESeq2 | NotchBio |
|---|---|---|
| Install PyDESeq2 | pip install pydeseq2 in clean env | Not required |
| Build counts matrix | Loop over quant.sf, concat, aggregate to gene | Automatic |
| Transpose to samples × genes | gene_counts.T (critical gotcha) | Not applicable |
| Build tx2gene mapping | Parse GENCODE GTF | Pre-loaded for GENCODE, Ensembl |
| Set reference level | Order contrast list: [var, test, ref] | Dropdown in UI |
| Multifactor design | design="~ batch + condition" | Checkbox per variable |
| Run deseq2() + Wald | Two method calls, multiprocessing setup | Automatic |
| apeGLM shrinkage | lfc_shrink() with coeff name | Always on |
| Symbol annotation | mygene query in batches | Included in output |
| Export DEG tables | pandas to_csv | Download from dashboard |
| Time to first results | 30-60 min including debugging | 10 min from quant.sf upload |
If you want to skip the counts matrix plumbing and environment management, notchbio.app runs PyDESeq2 and R DESeq2 interchangeably, with the same interface and the same annotated results table for either.
Further reading
Read another related post
How to Quantify RNA-Seq Reads with Salmon
Step-by-step Salmon tutorial: build a decoy-aware index, run salmon quant on paired-end reads, read the quant.sf output, and import into DESeq2 with tximport.
TutorialImport Salmon Output into R with tximeta and tximport
Import Salmon quant.sf into R with tximeta and tximport: build a tx2gene table, fix ID-mismatch errors, and set up a DESeqDataSet for multi-factor designs.
Research GuideWhy Cell Line RNA-Seq Experiments Fail
Passage drift, undetected mycoplasma, serum lot changes, and pseudoreplication silently corrupt cell line RNA-seq. What each looks like and how to prevent it.