How to Quantify RNA-Seq Reads with Salmon: Index, Quant, and Import to R
Most bulk RNA-seq pipelines still reach for alignment by default: STAR generates a BAM file, featureCounts tallies reads over exons, and the resulting matrix goes into DESeq2. That workflow is perfectly valid, but it takes tens of gigabytes of RAM, hours of wall time per run, and produces intermediate files you probably never open again. For the majority of standard differential expression studies, Salmon quantifies more accurately in a fraction of the time.
This tutorial walks through the complete Salmon workflow: building a decoy-aware transcriptome index, running quantification on paired-end FASTQ files, understanding what the output files contain, and importing everything into R for DESeq2. By the end, you will have a working pipeline you can drop into any project.
If you are still deciding whether Salmon is the right upstream tool, compare it with STAR vs HISAT2 vs Salmon: Which Aligner Should You Use? and keep Why Your Choice of Reference Genome Changes Your Results in mind when building the index. Once the quant.sf files exist, Importing Salmon Output into R: tximeta, tximport, and DESeq2 Setup is the next handoff.
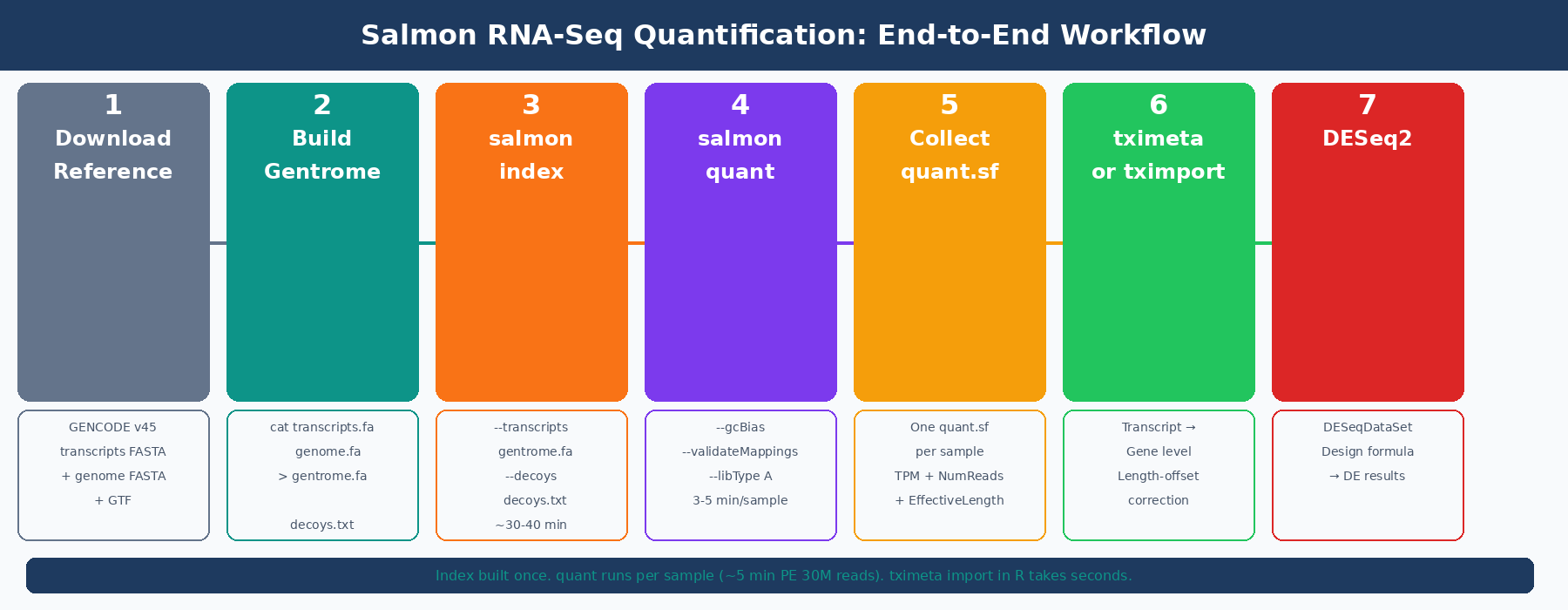
How Salmon Quantification Works (and Why It Is Faster Than Alignment)
Before running commands, it helps to understand what Salmon is actually doing, because it differs substantially from a genome aligner.
Traditional aligners like STAR find the exact genomic coordinates where each read maps. This requires a genome-scale index, a base-by-base dynamic programming alignment for each read, and a BAM file to store all the results. For a human sample, the index alone consumes about 38GB of RAM.
Salmon uses selective alignment: instead of computing precise genomic coordinates, it determines which transcripts in the reference are compatible with each read, then uses a statistical model to estimate abundances. The model accounts for GC content bias, positional bias, and fragment length distribution, all sources of systematic error that naive counting misses (Salmon documentation, COMBINE-lab). This produces transcript-level TPM values and estimated counts simultaneously, in minutes rather than hours.
The key constraint: Salmon only sees what is in its transcriptome index. It cannot detect novel splice junctions or transcripts missing from the annotation. For standard differential expression against a well-annotated organism, that is not a limitation. For experiments where novel isoform discovery is the goal, use STAR.
How to Install Salmon
Conda is the cleanest installation method. It handles all dependencies and keeps Salmon isolated from your system Python.
# Create a dedicated environmentconda create -n salmon -c conda-forge -c bioconda salmon -y
# Activateconda activate salmon
# Verifysalmon --version# salmon 1.10.0If you are on an HPC cluster, check for a module first:
module spider salmonmodule load salmon/1.10.0The Salmon GitHub releases page also provides pre-compiled binaries if conda is not available in your environment.
How to Build a Salmon Index (Decoy-Aware Method)
The index is built once per reference transcriptome version and reused across all samples. There are two ways to build it: a cDNA-only index (fast, small, slightly less accurate) and a decoy-aware index using the full genome (slower to build, more accurate, recommended for production use).
Why use a decoy-aware index? Reads from genomic regions that share sequence similarity with annotated transcripts can be spuriously mapped to those transcripts if the genome is not in the index. Including the genome as a decoy tells Salmon: if a read maps better to a genomic region than to any transcript, discard it rather than counting it. Accounting for fragments of unexpected origin can improve transcript quantification, particularly for gene families and pseudogenes (Salmon GitHub).
The nf-core/rnaseq pipeline uses the full genome as the decoy by default when running Salmon in pseudoalignment mode (nf-core docs).
Step 1: Download the Reference Files
For human GRCh38, use GENCODE for both the transcript FASTA and the genome FASTA, so chromosome naming is consistent (both use the chr prefix).
mkdir -p references && cd references
# Transcript FASTA (cDNA sequences)wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_45/\gencode.v45.transcripts.fa.gz
# Genome FASTA (primary assembly only, not patches or alt contigs)wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_45/\GRCh38.primary_assembly.genome.fa.gz
# GTF annotation (needed later for tx2gene mapping)wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_45/\gencode.v45.annotation.gtf.gzStep 2: Build the Gentrome and Decoy List
The decoy-aware index requires a single concatenated FASTA (transcripts first, then genome) and a file listing which sequences are the genomic decoys.
# Extract chromosome names from the genome FASTA for the decoys listgrep "^>" GRCh38.primary_assembly.genome.fa.gz \ | cut -d " " -f 1 \ | sed 's/>//' \ > decoys.txt
head -3 decoys.txt# chr1# chr2# chr3
# Concatenate: transcripts MUST come first, genome secondcat gencode.v45.transcripts.fa.gz \ GRCh38.primary_assembly.genome.fa.gz \ > gentrome.fa.gz
echo "Gentrome built."Transcript FASTA must come before genome FASTA in the gentrome
Salmon requires the transcriptome sequences to appear before the decoy (genomic) sequences in the concatenated file. Reversing the order causes the index build to fail or produce incorrect results. The decoys.txt file must also list only the chromosome names from the genome portion, not the transcript names.
Step 3: Build the Index
salmon index \ --transcripts references/gentrome.fa.gz \ --decoys references/decoys.txt \ --index references/salmon_index_hg38_v45 \ --gencode \ --threads 12
# --gencode strips the pipe-separated metadata from GENCODE transcript IDs# e.g. ENST00000456328.2|ENSG00000223972.6|... becomes ENST00000456328.2Index building for the full human genome takes roughly 30 to 40 minutes on 12 threads and requires around 32GB of RAM. The resulting index directory is about 20GB. This is a one-time cost: the index can be reused across every experiment that uses the same GENCODE release.
Lock your index to a specific GENCODE release version
The index encodes the transcript sequences at a specific annotation version. If you rebuild the index with a different GENCODE release mid-project, your expression estimates will be based on different transcript models and cannot be compared across samples. Build the index once, record the release in your methods, and do not update it during an experiment.

How to Run Salmon Quant on Paired-End RNA-Seq
With the index built, quantification runs per sample. For a 30-million-read paired-end library, a single Salmon quant run takes 3 to 5 minutes on 8 threads.
Single Sample
salmon quant \ --index references/salmon_index_hg38_v45 \ --libType A \ --mates1 data/raw/sample_ctrl1_R1.fastq.gz \ --mates2 data/raw/sample_ctrl1_R2.fastq.gz \ --threads 8 \ --gcBias \ --validateMappings \ --output results/salmon/sample_ctrl1_quantFour flags worth understanding:
--libType A tells Salmon to auto-detect strandedness from the data. Salmon will determine whether the library is unstranded (U), stranded forward (SF), or stranded reverse (SR) from the first 100,000 reads. The inferred library type is written to lib_format_counts.json in the output directory. You can also specify it directly: ISR for Illumina TruSeq stranded reverse, ISF for forward stranded.
--gcBias enables GC bias correction. Salmon estimates and corrects for the systematic tendency of GC-rich and GC-poor fragments to be sequenced at different rates. The DESeq2 documentation recommends using the --gcBias flag, which estimates a correction factor for systematic biases commonly present in RNA-seq data, unless you are certain your data do not contain such bias (DESeq2 vignette, Bioconductor).
--validateMappings enables selective alignment, which uses a chaining algorithm to score and validate mapping candidates rather than accepting the first compatible match. This has been the default behavior since Salmon 1.0.0, but specifying it explicitly ensures you get the most accurate results even if default behavior changes.
--seqBias can also be added to model sequence-specific bias (random hexamer priming effects). It adds a small amount of runtime and is worth enabling for datasets with known per-base content biases.
Automating Quantification Across All Samples
For a full experiment, use a bash loop or a simple Python subprocess wrapper:
#!/usr/bin/env bash# Usage: bash run_salmon.sh data/raw results/salmon
RAW_DIR="${1}"OUT_DIR="${2:-results/salmon}"INDEX="references/salmon_index_hg38_v45"THREADS=8
mkdir -p "${OUT_DIR}"
mapfile -t r1_files < <(find "${RAW_DIR}" -name "*_R1*.fastq.gz" | sort)
echo "Found ${#r1_files[@]} sample(s). Running Salmon quant..."
for r1 in "${r1_files[@]}"; do r2="${r1/_R1/_R2}" sample=$(basename "${r1}" | sed 's/_R1.*\.fastq\.gz//')
if [[ ! -f "${r2}" ]]; then echo " WARNING: R2 not found for ${sample}, skipping." continue fi
echo " Quantifying: ${sample}"
salmon quant \ --index "${INDEX}" \ --libType A \ --mates1 "${r1}" \ --mates2 "${r2}" \ --threads "${THREADS}" \ --gcBias \ --validateMappings \ --output "${OUT_DIR}/${sample}_quant" \ 2>> "${OUT_DIR}/${sample}_salmon.log"
done
echo "All samples complete."Run it as:
bash run_salmon.sh data/raw results/salmonEach completed sample produces its own directory under results/salmon/. Once all samples are done, run MultiQC to aggregate the Salmon logs into a single report:
multiqc results/salmon/ \ --outdir results/multiqc \ --filename salmon_multiqc_report \ --forceUnderstanding the Salmon quant.sf Output File
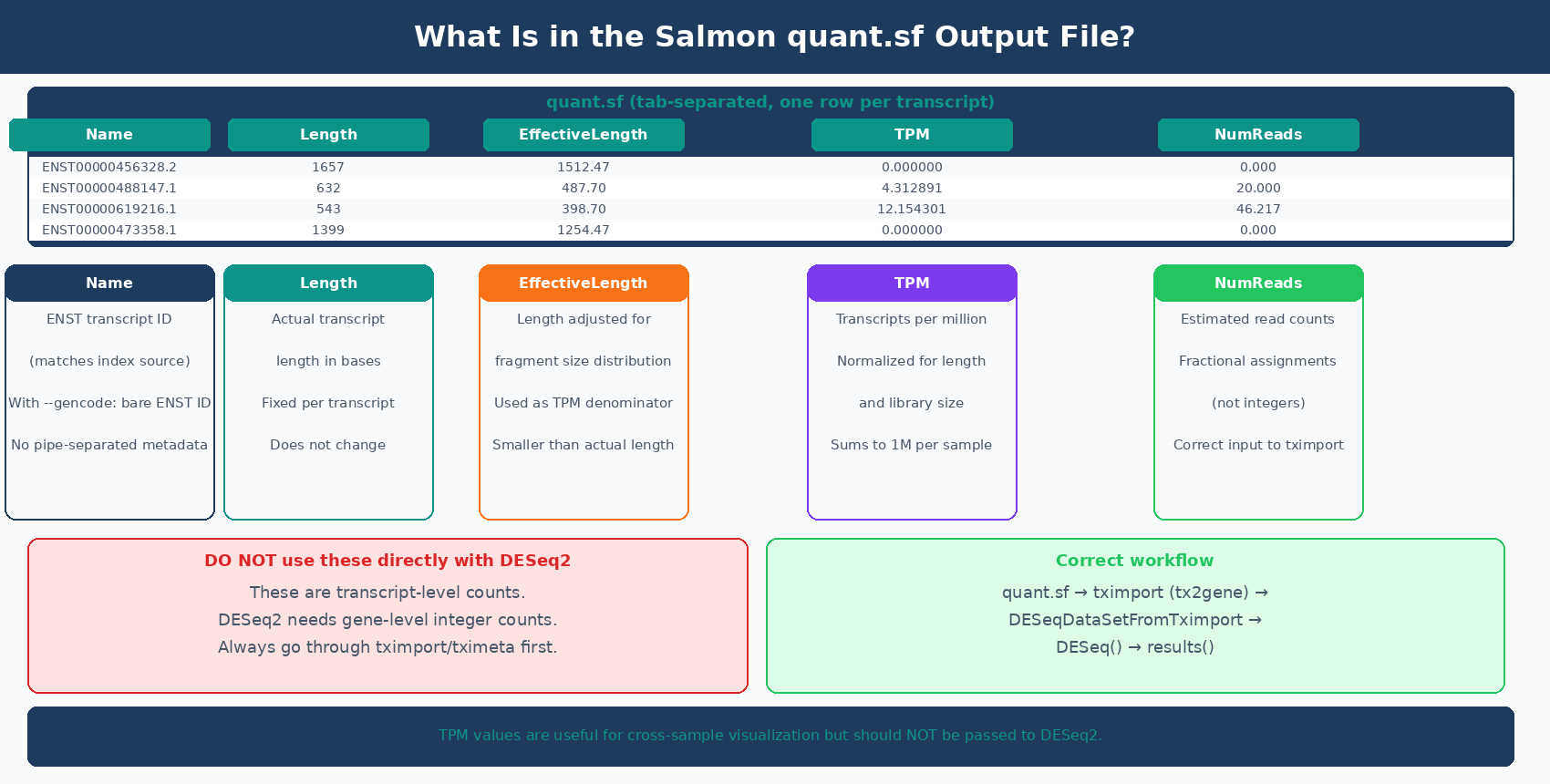
Each sample directory contains a quant.sf file: a tab-separated table with one row per transcript. Here is what each column means:
Name Length EffectiveLength TPM NumReadsENST00000456328.2 1657 1512.468 0.000000 0.000ENST00000488147.1 632 487.697 4.312891 20.000ENST00000619216.1 543 398.697 12.154301 46.217ENST00000473358.1 1399 1254.468 0.000000 0.000Name: the transcript identifier from your reference. With --gencode, this is the bare ENST ID without the pipe-separated metadata suffix.
Length: the actual length of the transcript in bases.
EffectiveLength: the length adjusted for fragment length distribution and positional bias. This is the denominator used in TPM calculation. Shorter fragments can only produce reads from a shorter effective window, so effective length is usually smaller than actual length.
TPM: transcripts per million. The number of reads originating from this transcript per million total reads, after normalization for effective length. TPM values sum to one million across all transcripts in a sample.
NumReads: the estimated number of reads originating from this transcript. These are probabilistic assignments, not integer counts: a read that is compatible with two transcripts contributes fractional counts to each. DESeq2 accepts and handles these fractional estimates correctly by rounding them internally.
Never use quant.sf NumReads directly with DESeq2 without going through tximport
The NumReads column contains transcript-level fractional counts. DESeq2 needs gene-level integer counts. Passing transcript counts directly to DESeq2 bypasses the aggregation and length-offset correction that tximport provides. The correct path is always: quant.sf → tximport/tximeta → DESeqDataSetFromTximport → DESeq2.
Checking Salmon Mapping Rates
Before importing results into R, check that your mapping rates are reasonable. The mapping rate is in the aux_info/meta_info.json file inside each sample directory:
import jsonimport osfrom pathlib import Path
results_dir = Path("results/salmon")sample_dirs = sorted(results_dir.glob("*_quant"))
print(f"{'Sample':<35} {'Mapped %':>10}")print("-" * 47)
for sample_dir in sample_dirs: meta_path = sample_dir / "aux_info" / "meta_info.json" if not meta_path.exists(): continue with open(meta_path) as f: meta = json.load(f)
sample_name = sample_dir.name.replace("_quant", "") pct_mapped = meta.get("percent_mapped", 0.0)
flag = " ✓" if pct_mapped >= 75 else " ⚠ LOW" print(f"{sample_name:<35} {pct_mapped:>9.1f}%{flag}")For a clean poly-A selected human or mouse RNA-seq library mapped to a GENCODE transcriptome, expect 75 to 92 percent mapping. A rate below 70 percent warrants investigation: wrong reference organism, genome-only reads (no cDNA-only reference), strandedness mismatch, or severe degradation are common causes.
MultiQC reads Salmon’s log files directly and surfaces the mapping rate per sample in its General Statistics table, so you rarely need to parse meta_info.json manually. The Python script above is useful when you want to integrate mapping QC into an automated pipeline or flag samples before running R code.
If you are not sure how to interpret the aggregated QC tables and per-sample warnings, Understanding Your QC Report: What FastQC and MultiQC Are Telling You is the companion guide.
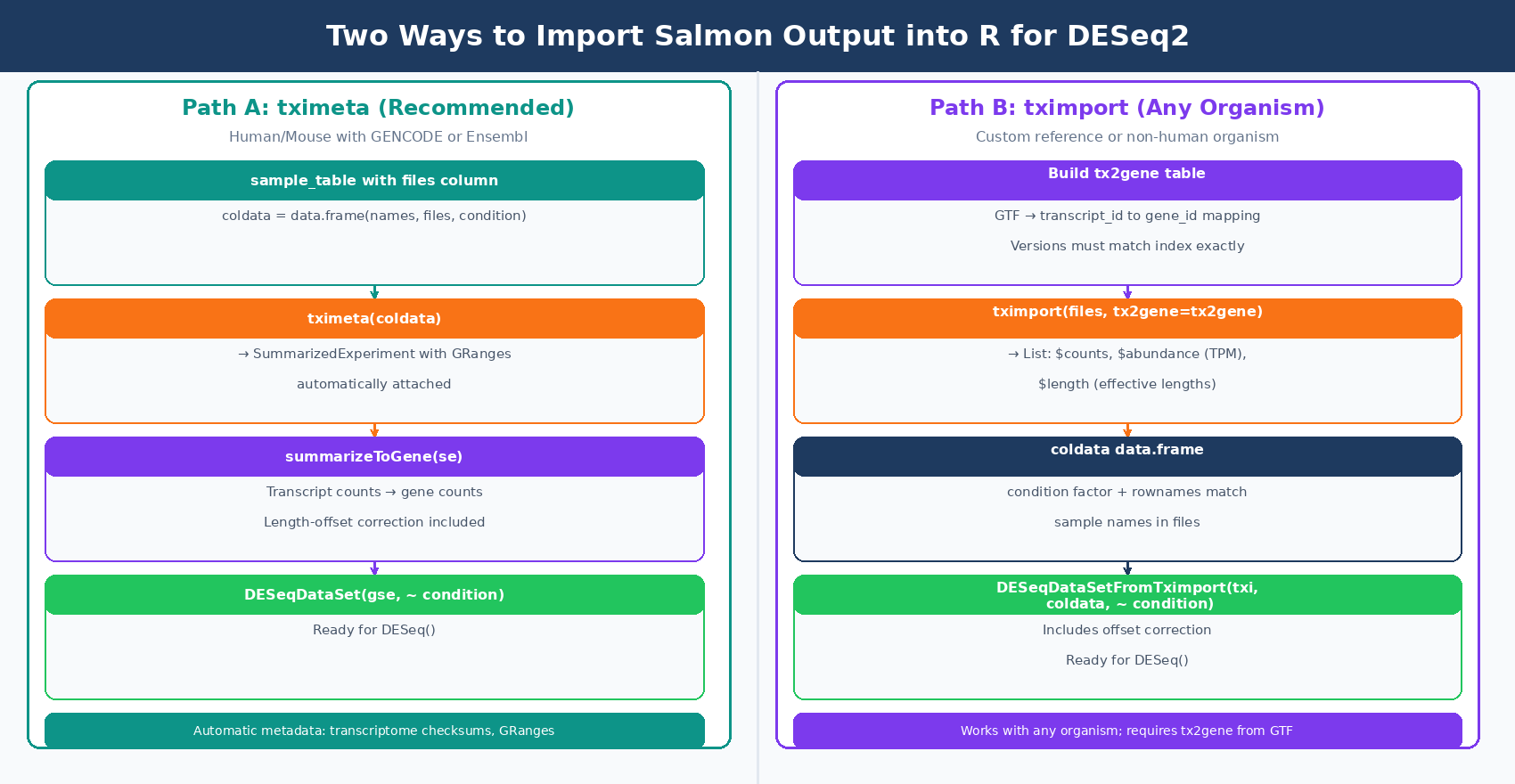
How to Import Salmon Output into R with tximeta
With all samples quantified and QC checked, import the quant.sf files into R. The recommended package is tximeta, which extends tximport by automatically attaching genome coordinate metadata when the transcriptome matches a known source (GENCODE, Ensembl, or RefSeq for human and mouse). Whereas tximport outputs a simple list of matrices, tximeta will output a SummarizedExperiment object with appropriate GRanges added (tximport vignette, Bioconductor).
For projects where tximeta does not recognize the transcriptome (custom references, non-human organisms), tximport is the fallback and works identically for the purposes of DESeq2 input.
Option A: tximeta (Recommended for Human/Mouse with GENCODE or Ensembl)
library(tximeta)library(DESeq2)
# Build a sample metadata table# Each row is one sample; 'files' column points to quant.sfsample_table <- data.frame( names = c("ctrl_1", "ctrl_2", "ctrl_3", "treat_1", "treat_2", "treat_3"), files = c( "results/salmon/ctrl_1_quant/quant.sf", "results/salmon/ctrl_2_quant/quant.sf", "results/salmon/ctrl_3_quant/quant.sf", "results/salmon/treat_1_quant/quant.sf", "results/salmon/treat_2_quant/quant.sf", "results/salmon/treat_3_quant/quant.sf" ), condition = factor(c("control", "control", "control", "treated", "treated", "treated")))
# Import with tximeta — automatically attaches GRanges metadata# if transcriptome is from GENCODE, Ensembl, or RefSeqse <- tximeta(sample_table)
# Summarize from transcript level to gene levelgse <- summarizeToGene(se)
# Build the DESeqDataSetdds <- DESeqDataSet(gse, design = ~ condition)
# Filter low-count genes before analysisdds <- dds[rowSums(counts(dds) >= 10) >= 3, ]
dim(dds)# Typically ~20,000 genes after filteringOption B: tximport (Works with Any Organism or Custom Reference)
library(tximport)library(DESeq2)
# Define sample filessample_names <- c("ctrl_1", "ctrl_2", "ctrl_3", "treat_1", "treat_2", "treat_3")
files <- setNames( file.path("results/salmon", paste0(sample_names, "_quant"), "quant.sf"), sample_names)
# Build the transcript-to-gene mapping from the GTF# You need this to aggregate transcript counts to gene levellibrary(GenomicFeatures)
txdb <- makeTxDbFromGFF( "references/gencode.v45.annotation.gtf.gz", format = "gtf")
k <- keys(txdb, keytype = "TXNAME")tx2gene <- AnnotationDbi::select(txdb, k, "GENEID", "TXNAME")
# Alternatively, build tx2gene from the GTF with bash:# zcat references/gencode.v45.annotation.gtf.gz | \# awk '$3=="transcript" {split($12,a,"\""); split($10,b,"\""); print a[2]"\t"b[2]}' \# > references/tx2gene.tsv
# Import: summarize transcript-level to gene-leveltxi <- tximport( files, type = "salmon", tx2gene = tx2gene)
# Sample metadatacoldata <- data.frame( condition = factor(c("control", "control", "control", "treated", "treated", "treated")), row.names = sample_names)
# Build DESeqDataSetdds <- DESeqDataSetFromTximport(txi, coldata, ~ condition)dds <- dds[rowSums(counts(dds) >= 10) >= 3, ]tx2gene version must match the FASTA version used to build the Salmon index
The transcript IDs in your GTF-derived tx2gene table must match the IDs in the quant.sf Name column exactly. If you built the index from GENCODE v45 transcripts, build tx2gene from the GENCODE v45 GTF. Mismatching versions causes silent ID lookup failures: many transcripts appear unmapped, your count matrix is mostly zeros, and tximport warns about unmatched transcripts without throwing an error.
Validating the Import: Checking Your Count Matrix
Before running DESeq2(), run three quick checks:
# 1. Confirm dimensions look reasonabledim(dds)# Rows: ~15,000-22,000 genes after filtering# Columns: number of samples
# 2. Check library sizes (should be in millions, reasonably balanced)colSums(counts(dds)) / 1e6# ctrl_1 ctrl_2 ctrl_3 treat_1 treat_2 treat_3# 23.1 22.8 21.4 19.7 24.3 22.1
# 3. PCA on variance-stabilized counts to confirm samples cluster by conditionvsd <- vst(dds, blind = TRUE)plotPCA(vsd, intgroup = "condition")# Controls should cluster together, treatments separately# If PC1 separates by batch instead of condition, add batch to the designA PCA that separates samples by condition rather than by extraction date or library prep run is your final confirmation that the import worked correctly and the experiment is worth proceeding with.
Running the Pipeline Without Writing It Yourself
The full workflow from FASTQ to differential expression, including Salmon quantification with a decoy-aware index, tximeta import, and DESeq2 analysis, is what NotchBio automates end to end. If you want to run this pipeline on your data without building the index, configuring the quant flags, or writing the R import code, upload your FASTQ files at notchbio.app and specify your organism and experimental design. The platform handles indexing, quantification, QC, and DE analysis, and returns the results with all parameters logged for reproducibility.
Further reading
Read another related post
The Reproducibility Crisis in Bulk RNA-Seq: What Actually Breaks
Half of published RNA-seq pipelines fail when someone else tries to run them. A practitioner view of what breaks and how to build for reproducibility.
TutorialDifferential Expression in Python with PyDESeq2: A Tutorial
PyDESeq2 brings DESeq2 statistics to Python. A complete tutorial covering model fitting, validation against R DESeq2, volcano plots, and enrichment export.
BioinformaticsWhy Reproducibility Should Not Be Optional in RNA-Seq Pipelines
Run snapshots, version pinning, and locked parameters should be the default, not a feature. A practitioner case for reproducibility-first RNA-seq platforms.