Why Your Choice of Reference Genome Changes Your Results
Two labs run the same RNA-seq experiment, using the same samples, the same aligner, and the same differential expression tool. They get different gene lists. Not slightly different. Meaningfully different: genes that appear significant in one analysis are absent from the other, and expression estimates for shared genes disagree by 50 percent or more in a non-trivial fraction of cases. The only difference between the two pipelines is which annotation database they used to build their reference.
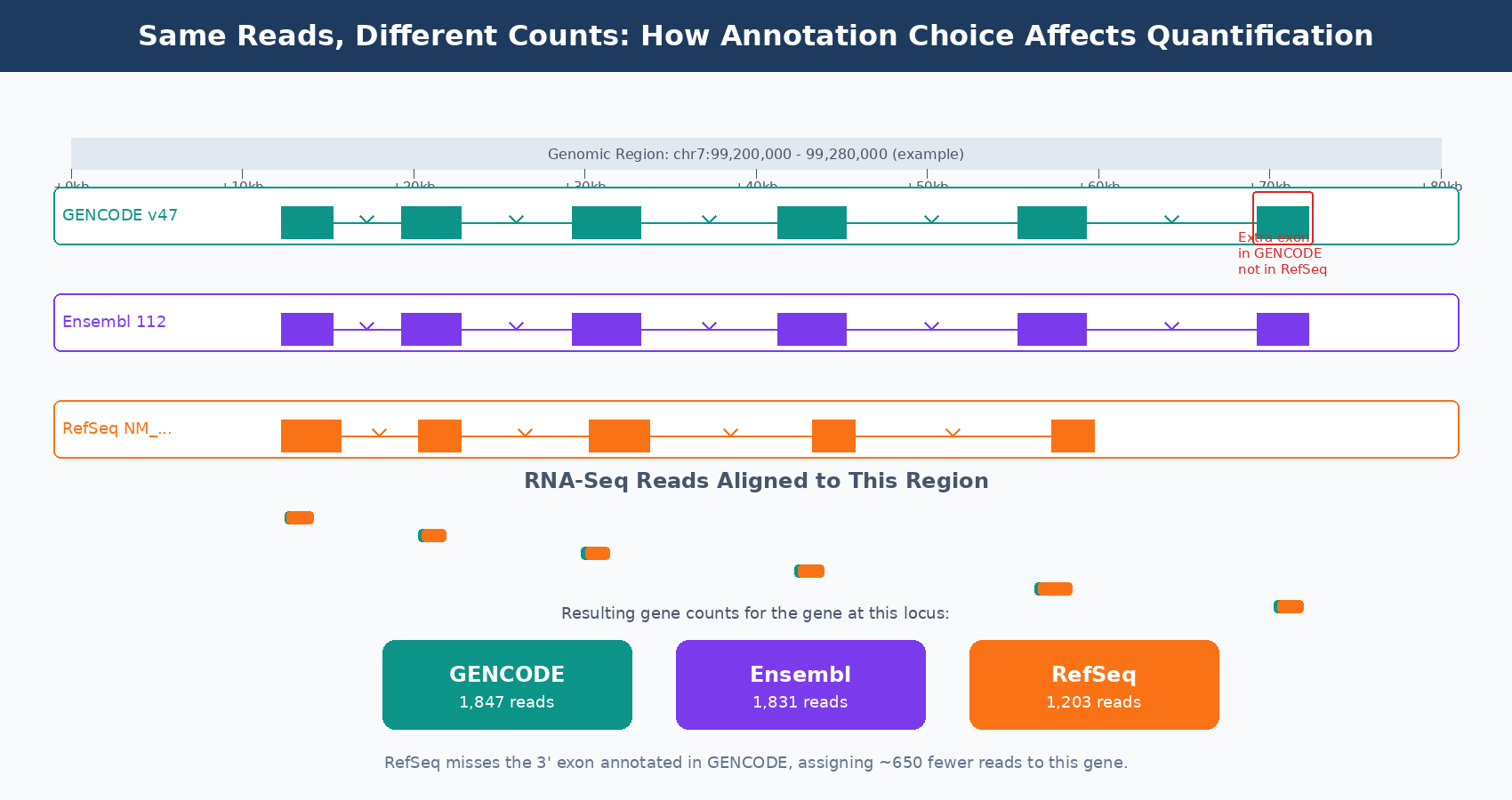
This is not a hypothetical. A systematic comparison of RNA-seq quantification across RefSeq, Ensembl, and UCSC annotations found that identical gene quantification results were obtained for only 16.3 percent of genes, roughly one in six. About 28 percent of genes showed expression level differences of 5 percent or higher between annotation databases, and for 9.3 percent of genes (over 2,000 genes), the relative expression levels differed by 50 percent or more (BMC Genomics, 2015). That is not annotation noise. That is a source of biological misinterpretation hiding inside a configuration choice that most protocols treat as a footnote.
This post explains what each annotation database actually contains, how those differences propagate into your read counts, and which choices to make for standard bulk RNA-seq differential expression analysis. For the rest of the computational context around alignment, quantification, and count generation, see Raw Reads to Counts: The Bulk RNA-Seq Pipeline Explained. For how those count differences affect downstream interpretation, read What Is a Count Matrix and Why Does It Matter.
The Annotation Problem in Plain Terms
A reference genome is just a DNA sequence: a FASTA file with the bases of each chromosome. It tells your aligner where reads can possibly map. The annotation, the GTF or GFF file, is the layer on top that tells your aligner and quantification tool where genes, exons, and splice junctions are located within that sequence. These are separate files that you must match carefully.
When you build a STAR index or run Salmon, you are encoding both the genome sequence and the annotation into a data structure that STAR or Salmon uses to make alignment decisions. The annotation determines which splice junctions are considered known, which affects how reads spanning exon-exon junctions are mapped. About 30 percent of junction reads fail to align without annotation assistance, while 10 to 15 percent map to alternative locations when the annotation changes. For non-junction reads, the choice of annotation matters much less: 95 percent of non-junction reads map to the same genomic location regardless of which annotation was used.
The real divergence happens at the quantification step. Once reads are aligned, the annotation determines which reads get assigned to which genes. When two databases define the boundaries of a gene differently, or include different isoforms, the same read may be assigned to different genes or counted in different genes entirely. That is where the expression estimate divergence comes from.
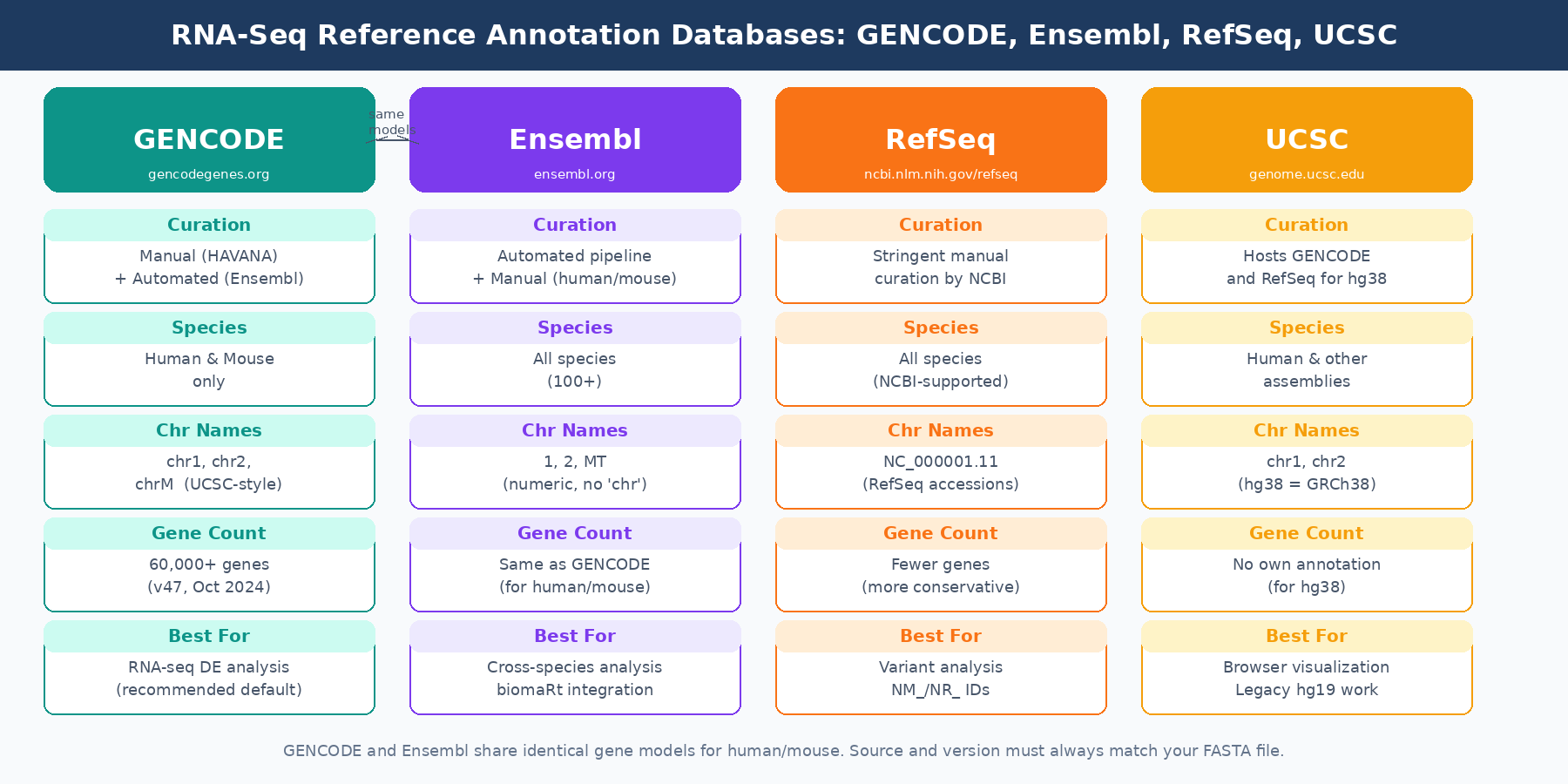
GENCODE: The RNA-Seq Default
GENCODE is the annotation most frequently used in RNA-seq analysis, and for good reason. It combines automated annotation from the Ensembl gene build pipeline with manual curation from the HAVANA team at the Wellcome Sanger Institute. Manual curation means human annotators review evidence from cDNA sequences, ESTs, proteins, and RNA-seq data to validate transcript models and add lncRNA and pseudogene annotations that automated methods miss.
GENCODE is focused specifically on human and mouse. As of GENCODE release 47 (October 2024), the human annotation covers over 60,000 genes including protein-coding genes, lncRNAs, pseudogenes, and small RNAs, across hundreds of thousands of transcript models (Nucleic Acids Research, 2025). The chromosome naming convention uses the UCSC-style chr prefix: chr1, chr2, chrM. This matters for compatibility with other tools.
For RNA-seq, the recommended file is the primary assembly annotation GTF, gencode.vX.primary_assembly.annotation.gtf.gz, which covers only the standard chromosomes and excludes patches and alternative haplotypes. Alternative haplotype regions introduce multi-mapping reads that inflate your multi-mapper rate without contributing useful signal.
The GENCODE website at gencodegenes.org provides downloads for all releases with clear version histories, making it straightforward to identify which release corresponds to which Ensembl release and ensuring reproducibility across experiments.
Download GENCODE primary assembly, not the comprehensive annotation
GENCODE offers two GTF files per release: the comprehensive annotation (all chromosomes, patches, haplotypes, scaffolds) and the primary assembly annotation (standard chromosomes only). For most RNA-seq workflows, use the primary assembly version. The comprehensive file adds complexity without meaningful benefit for differential expression analysis and increases your multi-mapping rate.
Ensembl: Same Gene Models, Different Format
Ensembl and GENCODE share the same underlying gene models for human and mouse. Since GENCODE version 3c, the GENCODE gene set corresponds to the Ensembl annotation, and each GENCODE release maps to a specific Ensembl release. The identifier, transcript sequences, and exon coordinates are nearly identical between equivalent versions.
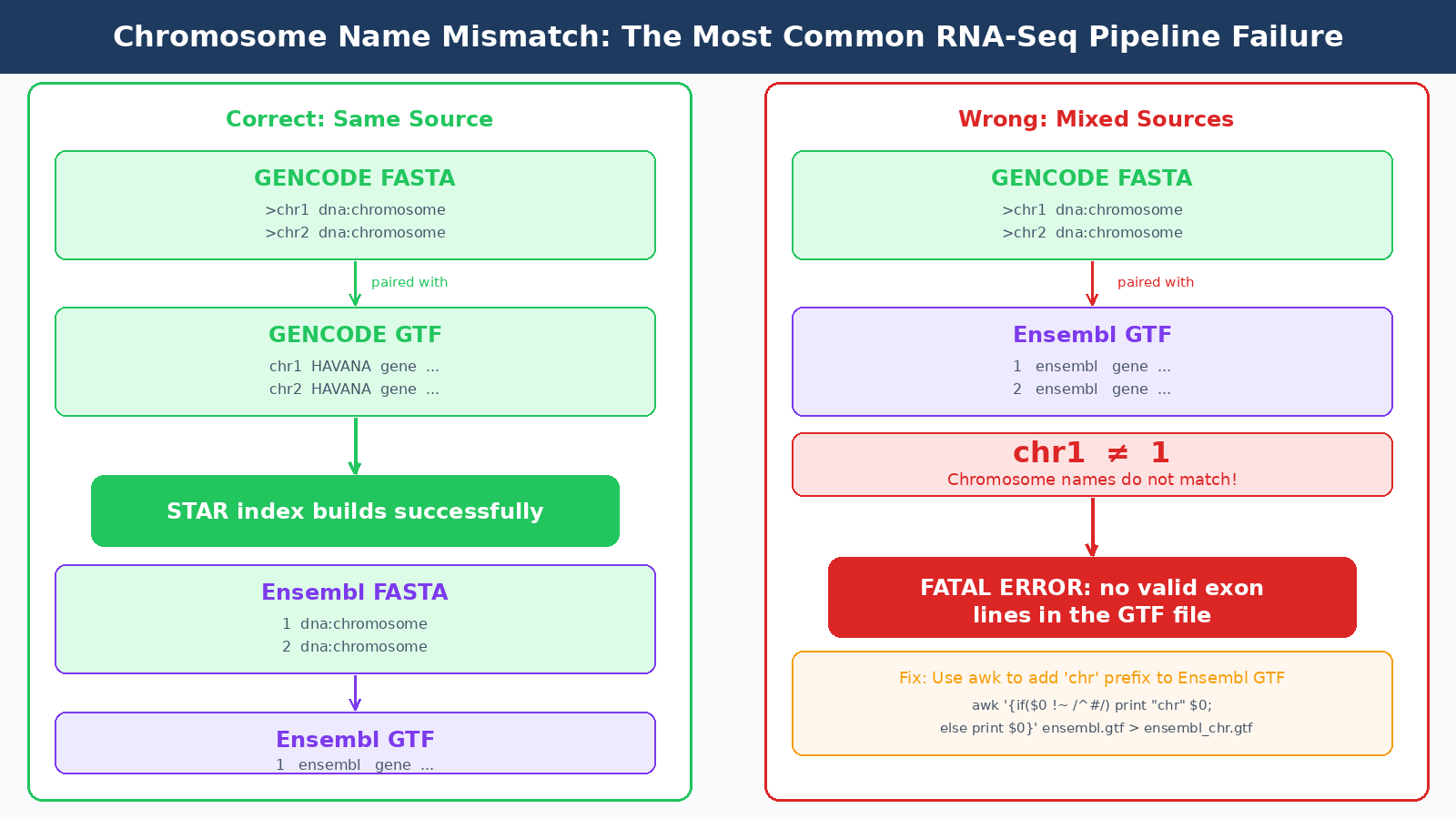
The meaningful differences are formatting. Ensembl uses numeric chromosome names without the chr prefix: 1, 2, MT. GENCODE uses chr1, chr2, chrM. This naming mismatch is one of the most common sources of the Fatal INPUT FILE error: no valid exon lines in the GTF file error in STAR. If your genome FASTA uses Ensembl-style names and your GTF uses GENCODE-style names, or vice versa, STAR cannot match chromosome names and your index build will fail or produce empty results.
The rule is simple: always download your FASTA and GTF from the same source and the same release. Ensembl FASTA with Ensembl GTF. GENCODE FASTA with GENCODE GTF. Mixing sources is the most common configuration mistake in RNA-seq pipeline setup, and it fails in ways that range from obvious (STAR exits with an error) to silent (STAR succeeds but a large fraction of reads map to the wrong location or nowhere).
Ensembl annotation files are available from the Ensembl FTP at https://ftp.ensembl.org/pub/release-{VERSION}/gtf/{species}/. For human GRCh38, the recommended file is Homo_sapiens.GRCh38.{VERSION}.gtf.gz.
RefSeq: More Conservative, Better for Variant Work
RefSeq is maintained by NCBI and applies more stringent curation criteria than Ensembl or GENCODE. A RefSeq transcript requires stronger experimental evidence to be included. This means RefSeq contains fewer transcripts and fewer genes overall: there are 21,958 common genes between RefGene, Ensembl, and UCSC annotations, but the databases diverge significantly beyond that shared core, with Ensembl annotating many more genes than RefSeq.
The tradeoff is quality versus completeness. RefSeq transcripts have a lower false-positive rate for novel or weakly supported isoforms, but they also miss a significant portion of the expressed transcriptome that Ensembl or GENCODE would capture. For differential expression analysis of protein-coding genes in well-characterized organisms, this difference is often small in practice. For lncRNA analysis or for organisms where manual curation is limited, it matters more.
RefSeq also has a formatting quirk: its transcripts have their own sequences that are independent of the genome assembly, meaning some variants may be present in RefSeq sequences that are not in the reference genome. This complicates mapping variant positions to transcript coordinates. For variant calling or allele-specific expression analysis, this is a genuine problem. For standard bulk RNA-seq expression quantification, it is usually irrelevant.
RefSeq is the dominant annotation in human genetics and variant databases. If your RNA-seq results need to be cross-referenced with ClinVar entries or variant effect predictions, using RefSeq consistently across your pipeline simplifies that integration. For pure expression analysis, GENCODE is generally the better starting point.
RefSeq transcript IDs start with NM_ or NR_; GENCODE IDs start with ENSG
Knowing your identifier namespace matters for cross-database lookups. RefSeq protein-coding transcripts begin with NM_, non-coding with NR_. GENCODE gene IDs begin with ENSG, transcript IDs with ENST. If a collaborator sends you a gene list from a different annotation source, you may need to convert identifiers using a tool like biomaRt or the Ensembl ID mapping service before the lists are directly comparable.
UCSC: A Host, Not a Builder
The UCSC Genome Browser is not primarily an annotation database in the same sense as Ensembl or RefSeq. For human GRCh38, UCSC hosts GENCODE annotations as its primary gene track and provides access to NCBI RefSeq coordinates. The older UCSC Genes track, also called Known Genes, was built using a proprietary gene predictor that is no longer in development and is not available for recent assemblies. If you encounter references to the UCSC Genes annotation in older protocols, they are describing this legacy track.
The practical implication is that for hg38 analysis, UCSC is not a distinct annotation source. If you download a GTF from the UCSC Table Browser for hg38, you are getting either GENCODE or NCBI RefSeq coordinates, not a UCSC-specific annotation. This was a source of confusion for years in the field: researchers would describe using the UCSC annotation without realizing they were actually using one of the other databases, just accessed through the UCSC interface.
One naming convention note: UCSC uses hg38 and mm10 for human and mouse assemblies where NCBI and Ensembl use GRCh38 and GRCm38 or GRCm39. These refer to the same underlying assemblies with minor versioning differences, but the naming discrepancy has historically caused researchers to mix incompatible FASTA files and GTF files when one source used one naming scheme and the other used another.
How Annotation Choice Affects Your Count Matrix
The practical effects of annotation divergence show up in four specific ways:
Multi-mapping rates. Databases that annotate more isoforms and pseudogenes, particularly Ensembl compared to RefSeq, have higher multi-mapping rates because reads from gene family members have more candidate loci to match. This is not necessarily wrong; Ensembl is capturing more of the transcriptome. But it does change how many reads get discarded as multi-mappers when you use a tool like STAR with default --outFilterMultimapNmax 10.
Junction read mapping. Annotated splice junctions help STAR map reads that span exon-exon boundaries. Without annotation, about 30 percent of junction reads fail to align. The annotation’s junction set directly determines which splice patterns are considered known and how ambiguous junction reads are resolved. An annotation that misses a highly expressed isoform will cause reads from that isoform to be unmapped or misaligned.
Gene count differences. The most direct effect: genes that are in one database but not another simply will not appear in your count matrix. For downstream GO enrichment or pathway analysis, a missing gene is invisible. If your biological question involves a gene that RefSeq has but GENCODE categorizes as a pseudogene, you will not see it in a GENCODE-based analysis.
Expression estimate divergence. When two databases define a gene’s boundaries differently, reads near the boundaries may be assigned to different genes. For genes with complex loci near overlapping features, this effect compounds. The 50 percent expression divergence observed for about 9 percent of genes in the BMC Genomics comparison is not random: it concentrates in genes with complex isoform structures and those in regions where annotations differ substantially between databases.
The Version Problem Is as Bad as the Source Problem
Choosing GENCODE over RefSeq is a decision you make once per project. The version problem is subtler and more insidious. Annotation databases release updates continuously, and each release changes gene coordinates, adds new transcripts, reclassifies gene biotypes, and sometimes changes gene boundaries or removes genes entirely.
If you build your STAR index using GENCODE release 43, run Salmon against a transcriptome built from GENCODE release 45, and try to merge count matrices from samples processed at different times with different versions, your gene IDs will partially mismatch and your expression estimates will be based on different gene models. This is not theoretical. Labs that process samples over months or years without version-locking their reference files accumulate this problem silently.
The practical solution is to version-lock everything at the start of a project. Record which GENCODE release, which Ensembl release, which genome assembly version, and which aligner version you used. Store the GTF and FASTA files used to build your index. Do not update the reference mid-project unless you have a specific, documented reason, and if you do, reprocess all samples from raw FASTQ files with the new reference.
Never mix annotation versions within a single experiment
If sample batch A was processed with GENCODE v43 and batch B with GENCODE v47, your combined count matrix will contain gene IDs that appear in one batch but not the other, and expression estimates that are not directly comparable. Gene annotation updates can move exon boundaries, reclassify lncRNAs as protein-coding, or add new genes entirely. Always process all samples in an experiment with the same reference files.
The Chromosome Name Compatibility Checklist
Most pipeline failures related to reference files come down to three checklist items that are easy to verify before you run anything expensive:
| Check | Command | What to Look For |
|---|---|---|
| FASTA chromosome names | grep ">" genome.fa | head -5 | chr1 (UCSC/GENCODE) or 1 (Ensembl) |
| GTF chromosome names | cut -f1 annotation.gtf | sort -u | head -10 | Must match FASTA format exactly |
| GTF has exon features | grep -m 5 "exon" annotation.gtf | Confirms GTF contains exon lines STAR needs |
| Versions match | Check filenames | Same release number in FASTA and GTF filenames |
Running these four checks before building your STAR index takes about 30 seconds and saves hours of debugging silent alignment failures.
Which Annotation to Use: A Practical Recommendation
For standard bulk RNA-seq differential expression analysis of human or mouse samples, GENCODE is the right default. It provides the most comprehensive annotation with both automated and manual curation, has clear versioning and download access, and is what STAR’s own documentation recommends for optimal compatibility. The UCSC genome browser FAQ also notes that high-throughput sequencing data results, including RNA-seq, are often using Ensembl/GENCODE annotations.
Use Ensembl directly if your pipeline requires Ensembl-style chromosome names (numeric without chr prefix), if you are working in a workflow that interfaces with Ensembl’s REST API or biomaRt lookups, or if consistency with a collaborator’s existing pipeline requires it. Since the gene models are the same, results are directly comparable between GENCODE and Ensembl releases of the same version.
Use RefSeq if your downstream analysis requires cross-referencing with NCBI variant databases, if you are doing allele-specific expression analysis, or if a specific journal or regulatory context requires RefSeq identifiers.
Regardless of which source you choose: lock the version at project start, download FASTA and GTF from the same source, verify chromosome name compatibility before indexing, and document the exact release in your methods section. These are the habits that make results reproducible by collaborators and by your future self.

Handling reference genome selection, versioning, and compatibility is one of the configuration steps that NotchBio automates. When you upload your FASTQ files and specify your organism and assembly version, the platform selects the appropriate GENCODE annotation, verifies compatibility, and locks the reference for the full run. If you want to skip the file management overhead and focus on results, notchbio.app handles the reference layer for you.
Further reading
Read another related post
How to Build a Salmon Index and Quantify Bulk RNA-Seq Reads
Step-by-step Salmon tutorial: download GENCODE references, build a decoy-aware index, run salmon quant with gcBias and seqBias on all samples, and verify mapping rates before DESeq2.
TutorialHow to Run FASTQ Quality Control with FastQC, fastp, and MultiQC
Full pipeline tutorial for bulk RNA-seq QC: run FastQC on raw reads, trim adapters with fastp, rerun QC, and aggregate reports with MultiQC. Includes parallel processing and how to read results.
TutorialHow to Download RNA-Seq Data from GEO and SRA Using sra-tools and pysradb
Step-by-step tutorial for downloading bulk RNA-seq FASTQ files from GEO and SRA. Covers prefetch, fasterq-dump, pysradb metadata extraction, batch downloads, and fixes for common errors.