Download RNA-Seq Data from GEO and SRA with sra-tools
Most RNA-seq projects start the same way. You find a published paper, open the GEO record, and now you need to pull down 24 FASTQ files.
The NCBI website gives you a link, but not one you can wget. The metadata is scattered across GSE, GSM, SRP, SRX, and SRR identifiers that the paper does not fully list. A graduate student’s first download attempt usually eats a full day.
This post shows you the fast way. Find the accessions, extract the sample metadata with Python, and download all FASTQ files in a single batch script.
We use the environment from the Ubuntu and macOS setup guide so sra-tools, pysradb, and GNU parallel are already installed.
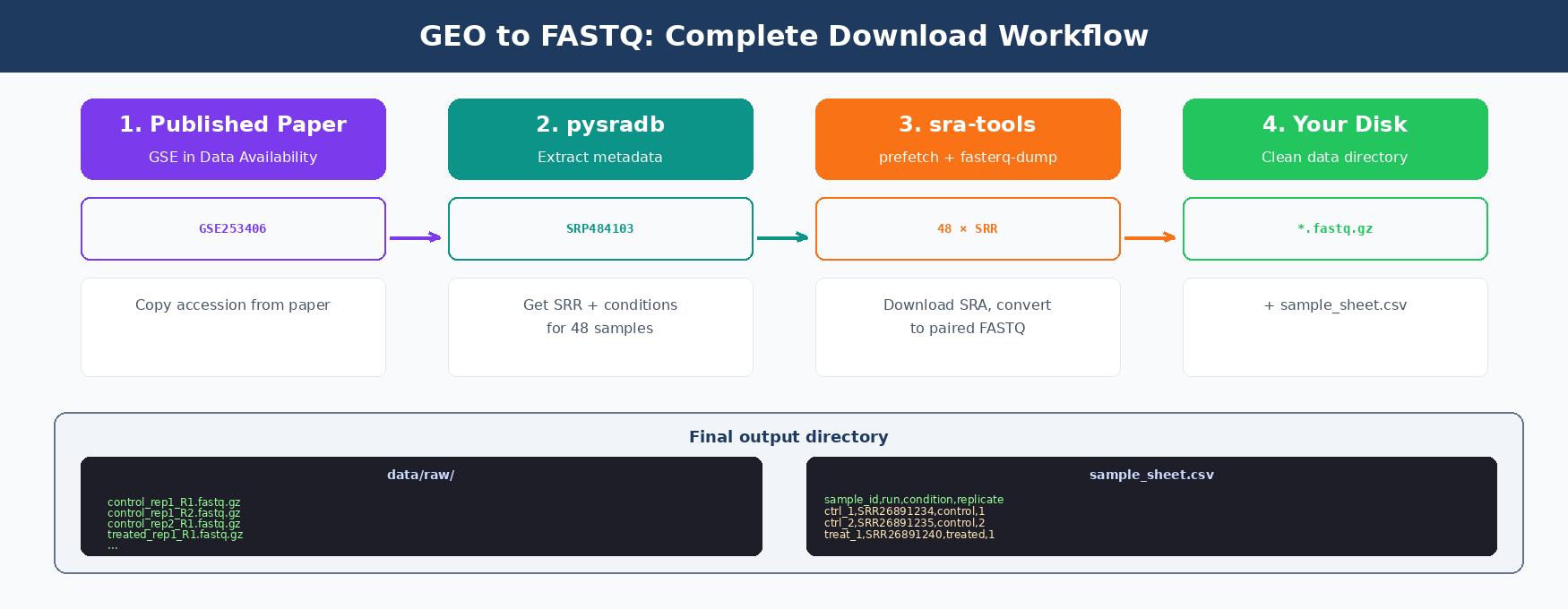
How GEO and SRA Accessions Work
Public sequencing data lives in two overlapping databases. GEO (Gene Expression Omnibus) is the user-facing archive. SRA (Sequence Read Archive) holds the actual raw reads.
Every study gets assigned a GEO accession starting with GSE. Individual samples get GSM IDs. The raw FASTQ files live under the SRA side, under SRP (study), SRX (experiment), and SRR (run) accessions.
You usually start from a GSE. You need to end up with SRR IDs, because those are what sra-tools downloads.
| Accession | What It Is | Example |
|---|---|---|
| GSE | GEO study (a paper’s entire dataset) | GSE253406 |
| GSM | GEO sample (one biological sample) | GSM8069123 |
| SRP | SRA study (maps 1:1 to GSE) | SRP484103 |
| SRX | SRA experiment (one library prep) | SRX22712345 |
| SRR | SRA run (the actual FASTQ file) | SRR26891234 |
| PRJNA | BioProject (NCBI wrapper for a study) | PRJNA1058002 |
One GSM can have multiple SRRs
A single sample (GSM) is sometimes sequenced across multiple lanes or flow cells. Each run becomes a separate SRR. When you build your sample sheet, you may need to merge multiple SRR files back into one sample.
How to Find RNA-Seq Datasets on GEO by Accession Number
Published papers usually mention the GSE in the Data Availability section.
Let’s use a real example. The paper “Integrative analysis of human plasma transcriptomics” lists GSE253406 as the accession.
You can visit the GEO record directly.
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE253406This shows you the sample names, sequencing platform, and library strategy. But it does not give you a one-click download for the FASTQ files.
For that, we need to get from GSE to the underlying SRRs. Two ways to do this.
Option 1: SRA Run Selector. Visit the SRA Run Selector for the study. The URL pattern is:
https://www.ncbi.nlm.nih.gov/Traces/study/?acc=GSE253406Click “Accession List” and you get a text file of SRR IDs.
Option 2: pysradb. The programmatic way. Faster and scriptable.
How to Extract Sample Metadata from GEO with pysradb in Python
pysradb is a Python package that queries SRA and GEO metadata from the command line and as a Python library. Two APIs: CLI and Python.
If you followed the Ubuntu and macOS setup guide, pysradb is already in your rnaseq environment. If not, install it now.
conda activate rnaseqpip install pysradbThe fastest CLI path: GSE to SRR in one command
pysradb gse-to-srr GSE253406Output:
study_alias experiment_alias run_accessionGSE253406 GSM8069123 SRR26891234GSE253406 GSM8069124 SRR26891235GSE253406 GSM8069125 SRR26891236...Save it to a file:
pysradb gse-to-srr GSE253406 --saveto GSE253406_srr.tsvGet detailed metadata including conditions
The gsm-to-srr output is just IDs. For a real experiment you want conditions, cell types, and tissue info. Use the metadata subcommand with --detailed:
# First convert GSE to SRPpysradb gse-to-srp GSE253406# GSE253406 SRP484103
# Then pull full metadata for the SRPpysradb metadata SRP484103 --detailed --saveto GSE253406_metadata.tsvThe output has columns for experiment_title, organism_name, library_strategy, instrument, run_accession, sample_attribute, and more. This is your sample sheet starting point.
The Python API for flexible metadata handling
For anything beyond a one-off download, use the Python API. You get a pandas DataFrame back, which is much easier to filter and transform.
from pysradb.sraweb import SRAwebimport pandas as pd
db = SRAweb()
# Get the full metadata table for a studymetadata = db.sra_metadata("SRP484103", detailed=True)print(metadata.shape)# (48, 28)
print(metadata.columns.tolist())# ['study_accession', 'experiment_accession', 'sample_accession',# 'run_accession', 'experiment_title', 'library_strategy',# 'organism_name', 'sample_attribute', ...]
# Filter to only RNA-seq runs (some studies mix assays)rna_only = metadata[metadata["library_strategy"] == "RNA-Seq"]
# Extract condition from sample_attribute (format: "source_name: ... || treatment: ...")rna_only["condition"] = rna_only["sample_attribute"].str.extract( r"treatment:\s*([^|]+)")
# Build a clean sample sheetsample_sheet = rna_only[[ "run_accession", "experiment_title", "organism_name", "condition"]].rename(columns={"run_accession": "sample_id"})
sample_sheet.to_csv("sample_sheet.csv", index=False)Always generate a sample sheet before downloading
Downloading 48 FASTQ files with filenames like SRR26891234.fastq.gz is painful to work with. A sample sheet that maps SRR IDs to meaningful names (sample_id, condition, replicate) makes the rest of your pipeline much easier. Build it now, not after you already have the files.

How to Download FASTQ Files with prefetch and fasterq-dump
The NCBI SRA Toolkit has two tools for this. Use them in order. Do not skip prefetch.
prefetch downloads the compressed SRA file. It is resumable, handles network hiccups, and is faster than alternatives.
fasterq-dump then converts the SRA file to FASTQ. It is multithreaded, unlike the older fastq-dump.
First-time setup for sra-tools
Configure the cache directory and output settings:
vdb-config --cfgThis opens an interactive menu. Set your cache directory to somewhere with plenty of disk space. A typical RNA-seq run is 2-5 GB as SRA, 5-15 GB as paired FASTQ.
Or set it non-interactively:
vdb-config --prefetch-to-cwd # store SRA files in current working directoryvdb-config --cfg-dir ~/ncbi # config locationDownload one sample
Let’s download SRR26891234 as a test.
# Step 1: download the SRA fileprefetch SRR26891234
# Step 2: convert to paired-end FASTQfasterq-dump SRR26891234 --split-files --threads 4 --progress
# Step 3: compress (SRA tools produces uncompressed FASTQ by default)gzip SRR26891234_1.fastqgzip SRR26891234_2.fastqYou now have SRR26891234_1.fastq.gz and SRR26891234_2.fastq.gz. That’s one paired-end sample.
fasterq-dump is much faster than fastq-dump
The older fastq-dump is still everywhere in tutorials but it is single-threaded and slow. One benchmark showed prefetch + fastq-dump took 25 minutes while fastq-dump alone took 77 minutes for the same file. fasterq-dump with 4 threads typically finishes in 3-5 minutes. Always use fasterq-dump.
Flags worth knowing
fasterq-dump SRR26891234 \ --split-files \ # split paired-end into _1.fastq and _2.fastq --threads 8 \ # use 8 CPU threads for extraction --progress \ # show a progress bar --outdir data/raw/ \ # where to write output files --temp /tmp/fasterq # scratch space (SSD speeds this up)--split-files is critical for paired-end data. Without it, you get one interleaved file that most downstream tools cannot read.
How to Download Many RNA-Seq Samples in Parallel (Batch Script)
One sample is easy. 48 samples is where people get stuck.
Two options: sequential bash loop (simple, slow) or GNU parallel (fast, parallel). We use the parallel version.
Sequential version (simple, good for understanding)
Save this as download_sequential.sh:
#!/bin/bashset -euo pipefail
# Read SRR IDs from a one-per-line fileSRR_LIST="srr_ids.txt"OUTDIR="data/raw"mkdir -p "$OUTDIR"
while read -r srr; do echo "[$(date +%H:%M:%S)] Processing $srr"
# Download SRA file prefetch "$srr"
# Convert to paired FASTQ fasterq-dump "$srr" \ --split-files \ --threads 4 \ --outdir "$OUTDIR"
# Compress gzip "$OUTDIR/${srr}_1.fastq" gzip "$OUTDIR/${srr}_2.fastq"
# Clean up the .sra file to save disk rm -rf "$srr"done < "$SRR_LIST"
echo "Done. Files in $OUTDIR"Run it:
bash download_sequential.shThis downloads samples one at a time. For 48 samples at ~5 minutes each, that’s 4 hours.
Parallel version with GNU parallel
Save this as download_parallel.sh:
#!/bin/bashset -euo pipefail
SRR_LIST="srr_ids.txt"OUTDIR="data/raw"JOBS=4 # number of concurrent downloadsTHREADS=4 # threads per fasterq-dump
mkdir -p "$OUTDIR"
process_sample() { local srr="$1" local outdir="$2" local threads="$3"
echo "[$(date +%H:%M:%S)] Processing $srr" prefetch "$srr" fasterq-dump "$srr" \ --split-files \ --threads "$threads" \ --outdir "$outdir" gzip "$outdir/${srr}_1.fastq" gzip "$outdir/${srr}_2.fastq" rm -rf "$srr"}export -f process_sample
parallel -j "$JOBS" \ process_sample {} "$OUTDIR" "$THREADS" \ :::: "$SRR_LIST"Run it:
bash download_parallel.shWith 4 parallel jobs and 4 threads each, a 48-sample study finishes in about 1 hour instead of 4.
Do not max out parallel downloads
NCBI rate-limits aggressive clients. More than 4-6 concurrent prefetches from the same IP will get throttled or temporarily blocked. If you need to pull a huge dataset faster, use the EBI ENA mirror, which serves FASTQ directly without the SRA conversion step.
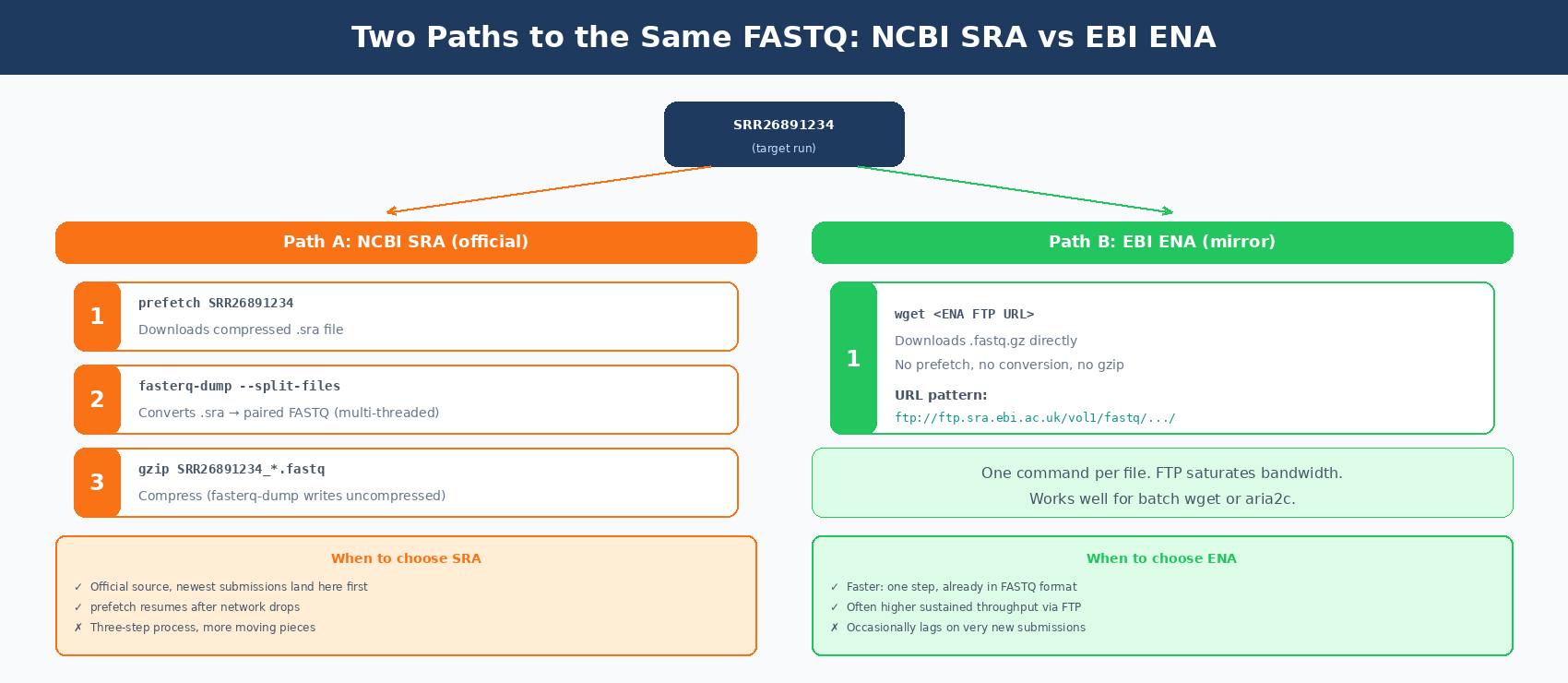
Using EBI ENA as a faster alternative
The European Nucleotide Archive mirrors SRA and serves FASTQ files directly. No prefetch step needed.
# ENA URL pattern: ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR268/091/SRR26891234/# Get the download URL with pysradb or construct it manually
# Using wget for one samplewget "ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR268/091/SRR26891234/SRR26891234_1.fastq.gz"wget "ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR268/091/SRR26891234/SRR26891234_2.fastq.gz"For studies with dozens of samples and limited bandwidth, ENA typically saturates your connection faster than the SRA route.
Common Download Errors and How to Fix Them
A few errors show up repeatedly. Fixes for each.
Error: “cannot resolve host”
NCBI requires outbound HTTPS on port 443. Most restrictive networks block this.
Fix: try from a different network (home wifi, university cluster), or use ENA which uses FTP on port 21.
Error: “prefetch is stuck”
prefetch downloads can hang on unreliable connections. Add a timeout and retry:
prefetch SRR26891234 --max-size 50g --resume yesThe --resume yes flag makes it pick up where it left off instead of restarting.
Error: “fasterq-dump: no rows matched”
Usually means prefetch did not finish and the SRA file is corrupt. Delete it and rerun:
rm -rf SRR26891234prefetch SRR26891234fasterq-dump SRR26891234 --split-filesError: “disk space full”
SRA files are large. A single human RNA-seq run is often 3-5 GB as SRA and 5-15 GB as uncompressed FASTQ. Always compress FASTQ as you go, and delete the .sra file after extraction:
rm -rf SRR26891234 # removes the directory prefetch createdA 48-sample study needs about 300-500 GB of scratch space during the download. Plan accordingly.
Error: “pysradb returns empty results”
Sometimes happens with very recent submissions that have not yet propagated to the SRAdb index. Two options: use the NCBI Entrez API directly via esearch/efetch, or wait 24-48 hours and retry.
# Fallback using Entrez utilitiesesearch -db sra -query "GSE253406" | \ efetch -format runinfo | \ cut -d',' -f1 | grep SRR > srr_ids.txtDo not manually rename FASTQ files before QC
SRA files have MD5 checksums. If you rename an SRR to something like control_1.fastq.gz before running FastQC, you lose the ability to verify the download. Keep SRR-named files until after QC, then rename (or use the sample sheet to alias them downstream).
Verify Your Downloads
Before moving on to QC, verify every file downloaded completely.
# Count expected vs actual filesexpected=$(wc -l < srr_ids.txt)actual=$(ls data/raw/*_1.fastq.gz | wc -l)echo "Expected $expected samples, got $actual"
# Check file sizes (a truncated download will often be < 100 MB)ls -lh data/raw/*.fastq.gz
# Spot check one file by peeking at the first readzcat data/raw/SRR26891234_1.fastq.gz | head -4# @SRR26891234.1 1/1# TCTTGGAAAGGCGCCTCCTCACA...# +# CCCCCGGGGGGGFGGGGGGGGGGG...If a file is suspiciously small or fails the read check, delete it and rerun prefetch + fasterq-dump for that single SRR.
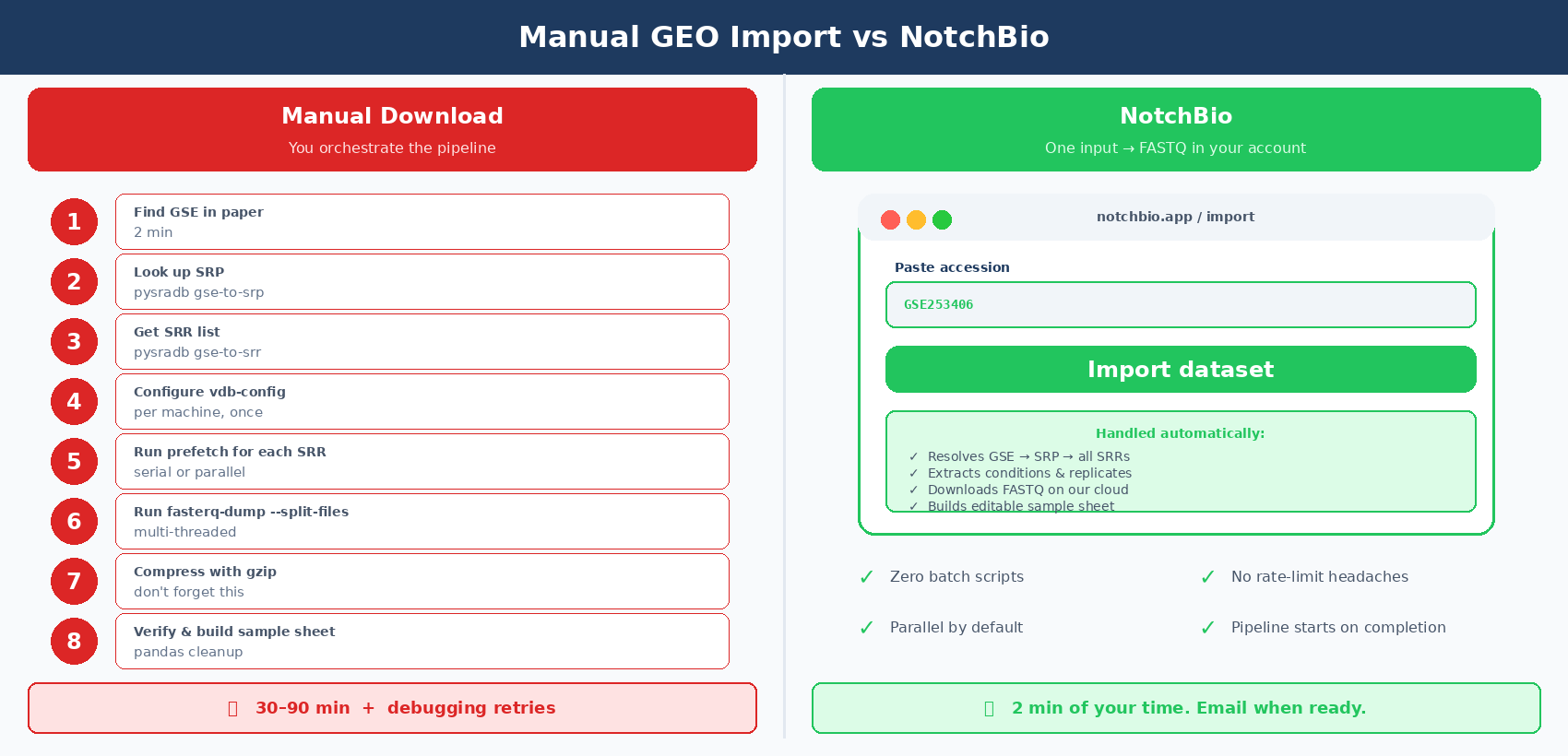
Manual SRA Download vs NotchBio: One-Click GEO Import
The workflow above is reliable but it is a lot of moving pieces. Accession hierarchy, vdb-config setup, prefetch and fasterq-dump flags, metadata parsing with pysradb, batch scripts, error recovery.
A 48-sample download takes around an hour of wall-clock time and maybe 30 minutes of active attention. Multiply that by every new public dataset you reprocess.
NotchBio imports public data directly. You paste a GSE or SRP accession, and the platform pulls the FASTQ files, extracts the metadata, and builds the sample sheet automatically. The pipeline starts running as soon as the download finishes.
| Step | Manual (sra-tools + pysradb) | NotchBio |
|---|---|---|
| Find SRP from GSE | pysradb gse-to-srp + manual check | Paste GSE, auto-resolved |
| Extract SRR list | pysradb gse-to-srr or Run Selector | Built in |
| Get sample metadata | pysradb metadata --detailed + pandas cleanup | Automatic, browsable table |
| Configure sra-tools | vdb-config per machine | Not required |
| Download FASTQs | prefetch + fasterq-dump + gzip | Cloud-side, parallel |
| Handle rate limits | You tune --jobs manually | Handled across accounts |
| Build sample sheet | Write pandas code for sample_attribute parsing | Auto-generated, editable |
| Resume failed downloads | Rerun script with --resume yes | Automatic retries |
| Time to first FASTQ in hand | 30-90 minutes for a 48-sample study | 5-15 minutes, in the background |
| Time you spend watching it | 30 minutes active + troubleshooting | 0 minutes, you get an email |
If you process a new GEO dataset more than once a month, the manual approach is fine. If you want the FASTQ files on disk in 10 minutes and move straight to QC, notchbio.app imports from GSE, SRA, or a list of SRR IDs directly.
Further reading
Read another related post
From Count Matrix to Volcano Plot: A DESeq2 Walkthrough in R
A complete DESeq2 tutorial in R: loading counts, building the design formula, running DE, applying lfcShrink, generating a volcano plot, and exporting results.
TutorialDESeq2 Contrasts: Multiple Conditions and Multi-Factor Designs
Three conditions, paired designs, two-factor experiments, and time courses: how to build the design formula, specify contrasts, and avoid common mistakes.
TutorialRNA-Seq Plots: Volcano, MA, and Heatmap in R and Python
Publication-ready RNA-seq plots in R and Python: volcano with ggplot2/ggrepel, MA plots, and DEG heatmaps with pheatmap and seaborn, plus 300 dpi export.