Why Deterministic Pipelines Beat AI-Generated Ones for RNA-Seq
The bioinformatics community has been running an uncontrolled experiment on AI-assisted analysis for the past two years. The results are in, and they are exactly what you would expect from an uncontrolled experiment: some things work remarkably well, some things fail in ways that are difficult to detect, and the failures tend to cluster in the places that matter most.
A senior bioinformatician described the pattern accurately in a widely discussed Reddit thread: you can get a generative AI to build an end-to-end genomics pipeline, but it is always broken, always in need of actual debugging, and it almost always produces results that require further investigation before you can trust them. That observation is not a dismissal of AI tools. It is a calibration. Understanding where AI assistance genuinely helps, and where deterministic systems are the only defensible choice, is the skill the field is now trying to develop.
What People Are Actually Using AI For
The community-level picture of AI use in bioinformatics is more nuanced than either the enthusiasts or the skeptics suggest. A thread on agentic coding agents for bioinformatics work, which generated significant engagement relative to its upvote count, revealed a consistent pattern across respondents.
Where AI assistance was working: installation and environment setup, writing scripts to submit jobs to a cluster, boilerplate file parsing and format conversion, generating plotting code from a described output, and explaining unfamiliar codebases. One researcher described using Claude Code to monitor a pipeline run on a cluster headnode, automatically fix trivial errors like wrong filepaths, and document the runs. For that class of task, agentic AI performs well.
Where it was struggling: the actual scientific analysis. One respondent ran AI-generated end-to-end RNA-seq notebooks and found the code “pretty good about writing end-to-end notebooks” but noted they still had to make “a few small fixes.” Another who built a website with AI assistance reported it was “ridiculously well” executed, then tried the same approach on a bioinformatics project and found it “performed surprisingly poorly,” setting up the wrong scope and loading unnecessary infrastructure.
The pattern is consistent. AI handles procedural and syntactic tasks well. It handles scientific judgment poorly.
Where AI Genuinely Works in a Bioinformatics Workflow
Being specific about where AI assistance adds value without introducing risk is more useful than a general endorsement or rejection.
Boilerplate and infrastructure code is the clearest win. Writing a SLURM submission script, setting up a conda environment file, generating a Makefile for a project directory structure, writing a parser for a file format you have never seen before: these tasks have a clear correct answer that an AI can produce reliably because they do not require scientific judgment about what the output should mean.
Plotting and visualization code is another area where AI assistance reliably saves time without introducing scientific risk. If you know what plot you want and can describe it, AI can write ggplot2 or matplotlib code faster than you can look up the syntax. The output is easy to inspect visually, so errors are immediately visible.
Explaining unfamiliar tools and codebases is underrated as an AI use case. Pasting a Nextflow process or an R function you do not understand and asking for an explanation, with the caveat that you will verify the explanation against the documentation, is low-risk and genuinely useful for accelerating comprehension.
Refactoring and documentation of code you have already written and understand is also reliable. AI can turn a messy analysis script into something more readable, add comments, and extract functions without changing the scientific content, because you can review the diff.
Where AI Fails, and Why the Failures Are Dangerous
The failures are dangerous not because they are obvious but because they are silent.
A bioinformatician who uses AI to write a DESeq2 analysis will get syntactically correct R code. The code will run. The output will look like a standard DESeq2 result. What the AI may have gotten wrong: the design formula, the reference level for the factor, whether lfcShrink was applied and with which estimator, whether the contrast was specified correctly for a multi-factor design, or whether the background gene set for enrichment analysis was the tested universe or the full genome. None of these errors produce an error message. All of them change the scientific result.
The same problem applies to parameter choices in upstream steps. An AI-generated alignment command for STAR will be syntactically valid. Whether --outSAMstrandField intronMotif should be included for a particular downstream tool, whether --quantMode GeneCounts is appropriate for the quantification approach being used, whether the --sjdbGTFfile matches the genome assembly being aligned to: these are choices that require knowing the experimental context, the downstream tool’s requirements, and the reference genome version. An AI without that context will make plausible-sounding choices that may be wrong.
The version problem compounds this. AI models have knowledge cutoffs. The DESeq2 or clusterProfiler API the model was trained on may not match the version installed in your environment. Package names get deprecated. Function signatures change. An AI that confidently generates code using a function that no longer exists in the current API is producing a failure mode that is harder to debug than a syntax error.
AI pipeline failures in bioinformatics are usually silent
Unlike syntax errors, scientific errors in AI-generated bioinformatics code do not produce error messages. A wrong design formula in DESeq2, an incorrect contrast specification, a mismatched reference genome, or a wrong background set in enrichment analysis will all produce output that looks valid. The only way to catch these is to understand the analysis well enough to know what correct output should look like.
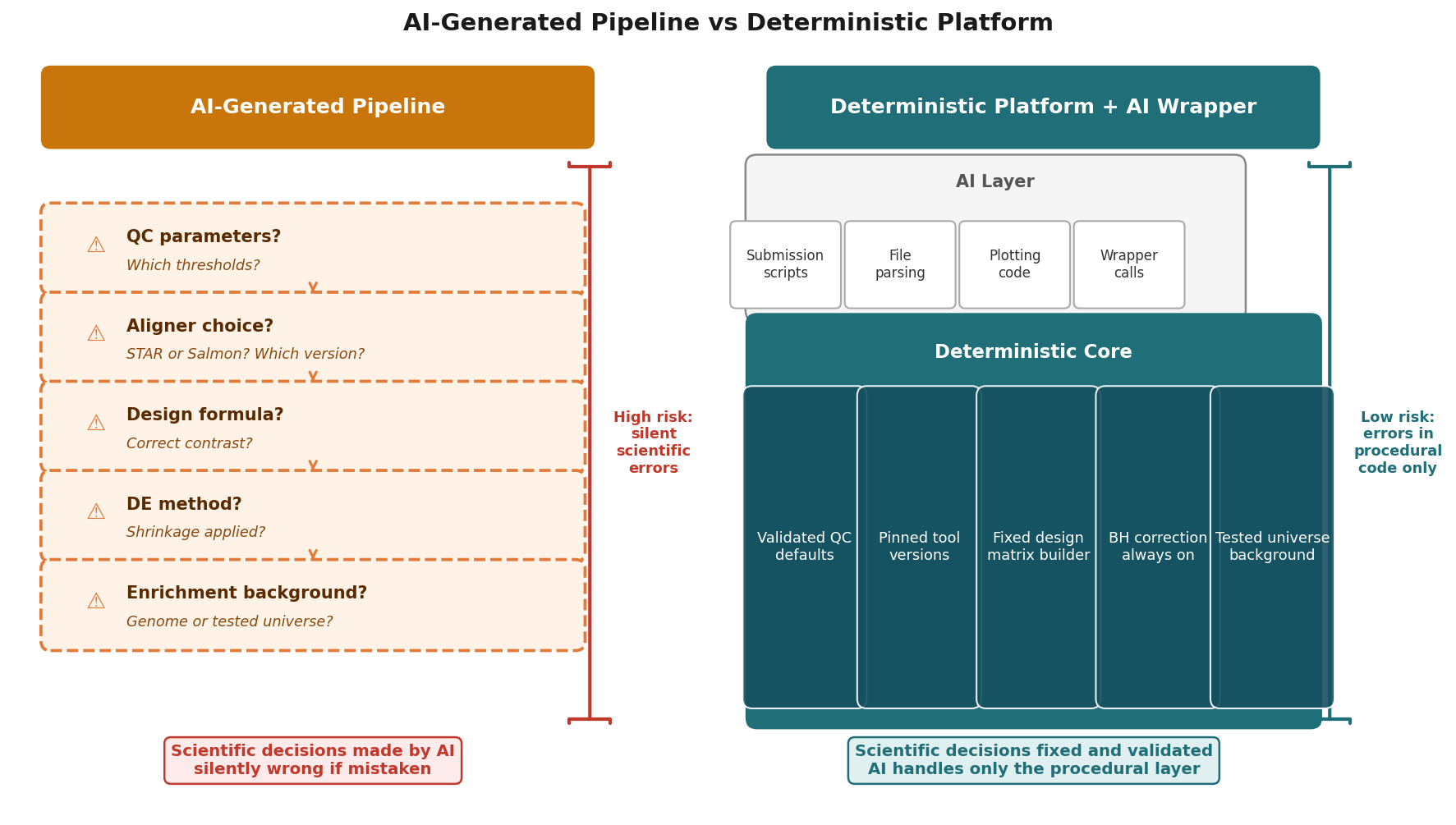
The Pattern That Actually Works: AI Calls Deterministic Pipeline
The most effective use of AI in bioinformatics is not AI generating the analysis. It is AI generating the code that orchestrates a deterministic analysis system.
Consider the difference between asking an AI to write a DESeq2 analysis from scratch and asking it to write a script that calls a validated API, uploads a count matrix, specifies a contrast, and retrieves the results. The first task puts scientific judgment in the hands of the model. The second task keeps scientific judgment in the hands of the validated system and uses the AI for what it is actually good at: writing procedural code around a well-defined interface.
This is not a hypothetical pattern. It is how production software engineering has used AI assistance from the beginning. You do not ask AI to design your database schema or choose your statistical model. You ask it to write the ORM layer or the plotting function after those decisions have been made by someone who understands the domain.
For RNA-seq specifically, the pattern means: the alignment tool, the quantification method, the normalization procedure, the statistical test, the multiple testing correction, and the enrichment analysis method are all fixed and validated. The AI writes the wrapper code, the submission scripts, the format conversions, and the visualization layers. The scientific core is deterministic and auditable.
How To Validate AI-Assisted Analyses Before You Trust Them
If you are using AI to assist with RNA-seq analysis and want to catch the failure modes described above, the following checks are not optional.
Run the analysis on a dataset you already understand. Before using AI-generated code on your real experimental data, run it on a public dataset with known results, such as the Pasilla dataset used in the Bioconductor DESeq2 vignette or the airway dataset. If the AI-generated code reproduces the published results on the canonical dataset, that is evidence the statistical approach is correct. If it does not, the error surface is clear.
Check every parameter against the current documentation. Do not assume the AI’s parameter choices are current. Open the tool documentation and verify that every argument the AI specified exists in the installed version and has the expected behavior. Pay particular attention to default values, which often change between major versions.
Verify the design formula by constructing it manually. Given your experimental groups, write out on paper what the design matrix should look like before running the code. Confirm that the AI-generated formula produces that matrix. DESeq2’s model.matrix() function lets you inspect the design matrix directly before any modeling occurs.
Check the enrichment background. Print out the length of the background gene list the AI passed to your enrichment function and confirm it matches the number of genes that were actually tested, not the size of the genome. A wrong background will produce inflated enrichment that passes all significance thresholds.
Ask the AI to explain its own choices. Prompting an AI to explain why it chose a particular contrast specification or enrichment method often surfaces uncertainty in the model itself. If the explanation contains hedges like “this approach is common” or “this should work for most cases,” treat those as signals that the AI is generalizing from training data rather than reasoning from your specific experimental context.
The Calibrated Position
The researchers who are using AI assistance most effectively in bioinformatics are not treating it as a replacement for domain knowledge. They are treating it the way an experienced programmer treats a junior developer: useful for getting things done faster, but requiring oversight proportional to the complexity of the task and the invisibility of the failure modes.
For the procedural layer of bioinformatics work, that oversight is light because the failures are visible. A broken submission script fails loudly. A malformed file format raises an exception. For the scientific layer, that oversight requires enough domain knowledge to know what correct output looks like, which means the researcher needs to understand the analysis even if they are not the one writing the code for it.
Let AI write your wrapper code, your submission scripts, your plotting functions, and your format converters. Let a validated, deterministic platform like NotchBio run your RNA-seq analysis, where the alignment parameters, normalization method, statistical test, and multiple testing correction are fixed, documented, and the same every time. The combination gives you the speed of AI assistance on the procedural layer and the reliability of a validated system on the scientific layer. That is a defensible workflow. Letting AI make unchecked scientific decisions on your experimental data is not.
Related Reading
Further reading
Read another related post
Import Salmon Output into R with tximeta and tximport
Import Salmon quant.sf into R with tximeta and tximport: build a tx2gene table, fix ID-mismatch errors, and set up a DESeqDataSet for multi-factor designs.
Research GuideWhy Cell Line RNA-Seq Experiments Fail
Passage drift, undetected mycoplasma, serum lot changes, and pseudoreplication silently corrupt cell line RNA-seq. What each looks like and how to prevent it.
Research GuideSTAR vs HISAT2 vs Salmon: Which Aligner Should You Use?
STAR aligns to the genome, HISAT2 uses less memory, Salmon skips alignment. What each approach means for your RNA-seq results and when each is the right call.