Nextflow vs No-Code Platforms: The Right Tool For Your Lab
Nextflow has won. A 2024 analysis of workflow management system citations found that Nextflow, with a citation share of roughly 43 percent in 2024, has become the main driver behind adoption of bioinformatics-based workflow management systems, and the nf-core community has built more than 100 production-grade pipelines on top of it. If you are building a bioinformatics core, contributing to large consortium analyses, or doing work that will be shared across institutions, the answer to “what workflow manager should I use” is almost certainly Nextflow.
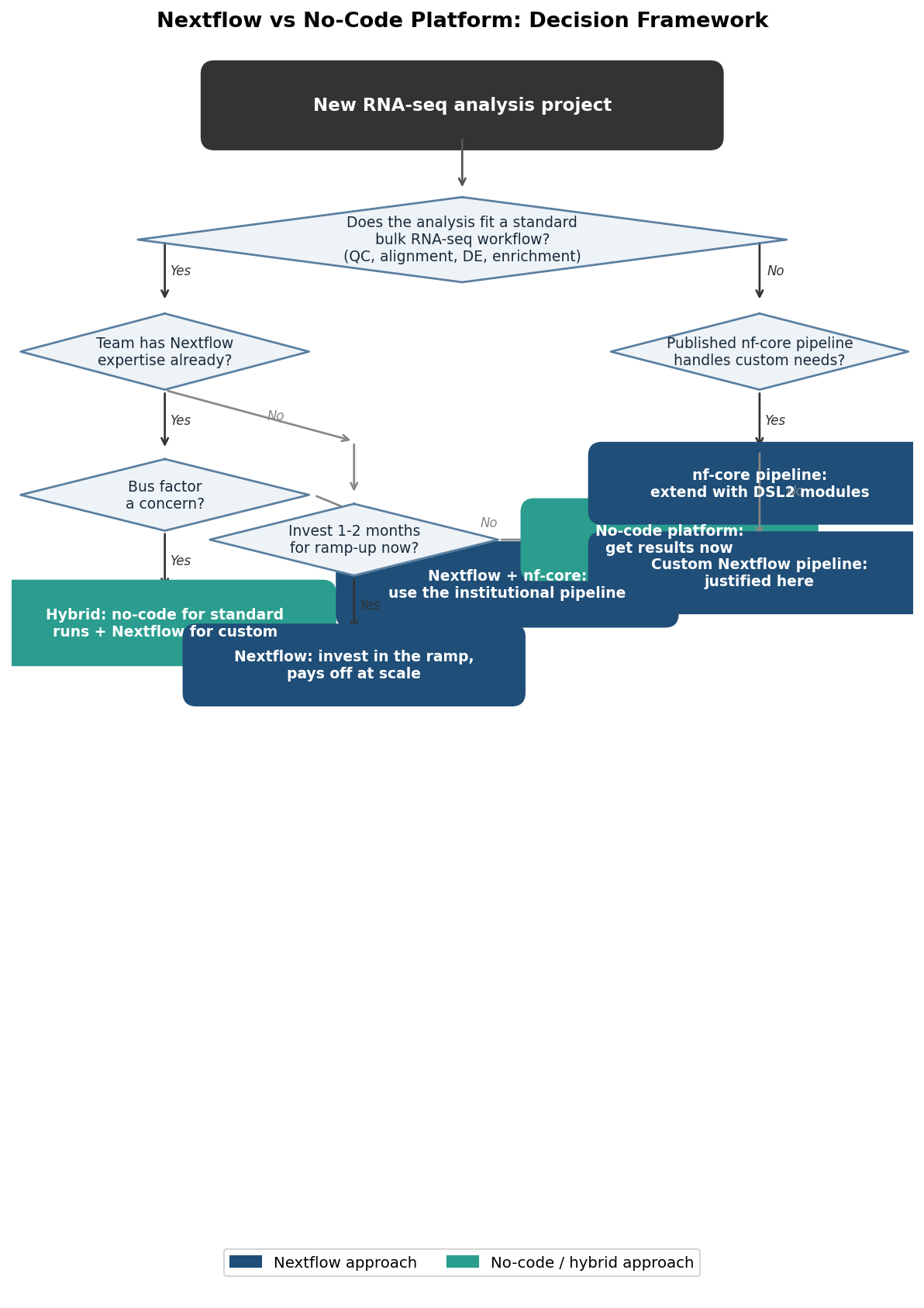
That is not the question most labs are actually asking. Most labs are asking a different question: should we invest three months in standing up a Nextflow infrastructure, or should we use something that works now? That question has a different answer, and pretending it does not is how labs end up with bioinformaticians spending their time on pipeline engineering when the biology is waiting.
This post argues honestly for both tools and gives you a framework to decide which fits your lab today.
What Nextflow Is Actually Good At
Nextflow’s genuine strengths are worth naming precisely because the case for it is sometimes overstated in ways that mislead labs with simpler needs.
Full control over every step. If your analysis requires a custom pre-processing step between alignment and quantification, a non-standard normalization approach, or a tool that is not in any existing pipeline, Nextflow lets you implement it exactly as you need it. No-code platforms cover what they were designed to cover. Nextflow covers whatever you can write.
Scaling across compute environments without rewriting. The same Nextflow pipeline runs on a laptop, a SLURM cluster, AWS Batch, or Google Cloud Life Sciences by changing a configuration profile. For groups that move analyses between local HPC and cloud, this portability is genuinely valuable and hard to replicate in other architectures.
Institutional pipeline sharing. The nf-core framework gives organizations a standard way to share, validate, and version-control pipelines. When six research consortia need to run the same RNA-seq analysis on different hardware in different countries, a shared nf-core pipeline is the right answer. The Harvard Chan Bioinformatics Core recognized the imperative of transitioning to nf-core as a result of its ability to handle reproducibility and scalability requirements at institutional scale.
Resume and fault tolerance at scale. Nextflow’s content-addressed work directory means that when a 200-sample pipeline fails on sample 147 because of a transient cluster error, you restart from sample 147, not from sample 1. For large-scale analyses where compute time is a real cost, this matters.
What Nextflow Costs
The same 2024 Seqera survey that documented Nextflow’s dominance also documented its learning curve. Every community thread on the subject acknowledges it openly: the median new user takes four to eight weeks before they are comfortable writing their own processes, wiring channels correctly, and debugging execution without help.
That ramp is not a permanent cost, but it is a real one. The question is who pays it, when, and whether it delivers value proportional to the investment.
A one to two month ramp for the first developer. Writing a minimal Nextflow pipeline, configuring it for your cluster, and debugging it to the point of reliable execution takes a dedicated developer approximately one to two months. If that developer is a bioinformatician who also needs to analyze data, that month comes from somewhere.
Ongoing maintenance. Nextflow pipelines require maintenance as tools update, as cluster configurations change, as container registries move images. In a well-resourced bioinformatics core, that maintenance is part of the job. In a small lab with one bioinformatician, it is a recurring drain on analysis time.
The bus factor. This is the question no one asks during the planning meeting.
Ask yourself: what happens when the Nextflow person leaves?
If your lab’s entire computational infrastructure depends on one person who knows how it works, your bus factor is one. When that person leaves for their next position, you are not just hiring a replacement. You are also paying the full ramp cost again while the replacement learns your specific pipeline configuration. No-code platforms reduce bus factor because the infrastructure is maintained externally and the interface is learnable in hours, not weeks.
The Nextflow expertise hiring problem. The jobs boards bear this out: Nextflow expertise is now explicitly requested in bioinformatics job listings in a way that Python or R expertise is not. Finding a developer who knows Nextflow well enough to maintain a production pipeline is harder than it was three years ago. The demand has grown faster than the supply of experienced users.
What No-Code Platforms Cover
The label “no-code” is slightly misleading. What these platforms actually do is embed code that has already been written, validated, and version-pinned into a workflow that a researcher accesses through an interface rather than a terminal. The code exists. You just do not have to write or maintain it.
For standard bulk RNA-seq, the coverage is comprehensive. Quality control with FastQC and MultiQC, adapter trimming with fastp, pseudoalignment with Salmon or alignment with STAR, quantification, import into DESeq2, differential expression with lfcShrink, enrichment analysis with clusterProfiler: these are the steps that appear in the majority of bulk RNA-seq papers. A well-built no-code platform handles all of them with validated defaults that reflect current best practices.
What no-code platforms cannot cover: custom pre-processing steps that do not fit a standard module, non-model organisms that require custom annotation handling, analyses that require the intermediate outputs of one step as non-standard inputs to another, and any novel method that was published after the platform was last updated.
The honest characterization is: no-code platforms cover 90 percent of standard bulk RNA-seq workflows. Nextflow covers 100 percent, including the custom 10 percent. The question is whether your lab’s 10 percent actually needs custom handling, or whether you are building infrastructure for hypothetical future needs while current analyses wait.
The Hybrid Pattern That Actually Works
The most effective setup for a research lab that has both standard analysis needs and occasional novel methodology is not “Nextflow for everything” or “no-code for everything.” It is a deliberate split.
Use a no-code platform as the production layer for all standard analyses. Every sample that goes through the standard bulk RNA-seq workflow (FASTQ quality check, alignment or quantification, differential expression, enrichment) runs through the platform. Results are reproducible, methods sections are auto-generated, and bioinformaticians are not interrupted for routine runs.
Reserve Nextflow for the genuinely custom work. The novel normalization approach, the multi-omics integration pipeline, the collaboration with an external consortium that requires nf-core standards. These are the analyses where the investment in Nextflow infrastructure is actually justified by the scientific requirement.
This split also resolves the bus-factor problem. The Nextflow expertise is concentrated in the people doing the genuinely novel work, which is the work where that expertise matters. The standard analyses do not depend on that expertise being present.
The Full Comparison
| Dimension | Nextflow | No-Code Platform |
|---|---|---|
| Initial ramp time | 4 to 8 weeks for a new developer | Hours to days for any researcher |
| Custom step support | Full: any tool, any step | Limited to what is built in |
| Cloud / HPC portability | Native: profile-based config | Depends on platform provider |
| Institutional sharing | nf-core framework, versioned modules | Varies by platform |
| Bus factor risk | High if one person owns the pipeline | Low: infrastructure maintained externally |
| Reproducibility | Excellent with correct config management | Excellent: locked runs by default |
| Maintenance overhead | Ongoing: tools update, clusters change | Low: maintained by provider |
| Standard RNA-seq coverage | 100% (including custom) | ~90% of standard workflows |
| Methods section generation | Manual or with nf-prov plugin | Automatic on most platforms |
| Cost | Compute cost only (open source) | Subscription or usage-based fee |
| Best for | Bioinformatics cores, consortium work, novel methods | Labs without dedicated pipeline engineers |
Making the Call
The right answer for your lab depends on three things you can assess in about fifteen minutes: what percentage of your analyses are standard bulk RNA-seq, whether you have someone who can own the Nextflow infrastructure without it becoming a distraction from analysis, and what the cost of a six-to-eight week ramp looks like given your current analysis queue.
If most of your work is standard, your team is small, and your queue cannot absorb a ramp period, start with a no-code platform. When you encounter the first analysis that genuinely requires custom Nextflow work, you will have a clearer picture of whether the investment is justified. If you are at an institution that already has Nextflow expertise and shared infrastructure, use it. The marginal cost of adopting an existing nf-core pipeline is low.
NotchBio is built specifically for the case where a no-code platform is the right answer today. Standard bulk RNA-seq from FASTQ to enrichment results, validated defaults, reproducible run records, auto-generated methods text, and no Nextflow knowledge required. When your analysis needs evolve past what a platform covers, the results you have run through NotchBio are fully exportable and compatible with any downstream analysis tool or workflow manager.
If you are evaluating whether NotchBio fits your current analysis needs, the fastest way to find out is to upload a sample metadata table and a set of FASTQ paths at notchbio.app and run the first analysis. The pipeline configuration is automatic and the run takes less time than reading this post.
Related Reading
Further reading
Read another related post
What Actually Happens to Your RNA Sample Before It Becomes Data
From tissue extraction to FASTQ file: a clear breakdown of RNA-seq library prep, sequencing chemistry, and what goes wrong at each step.
BioinformaticsWhen to Use edgeR vs DESeq2 vs limma-voom
DESeq2, edgeR, and limma-voom all test differential expression but use different models, normalization, and assumptions. Here is when each one wins.
Research GuideUnderstanding Your QC Report: FastQC and MultiQC
A module-by-module guide to reading FastQC and MultiQC output for RNA-seq data — what each plot means, which failures matter, and which you can safely ignore.