Self-Service RNA-Seq For Labs Without A Bioinformatician
The sequencing run completed three weeks ago. The FASTQ files are on your institution’s server. Your PI is asking for the DEG list. And the bioinformatician who was supposed to analyze it is finishing up a previous project that has run over schedule, is splitting time with two other groups, and cannot start on your data until next month.
This is not an unusual situation. It is the normal situation for a large fraction of wet-lab groups. The staffing model that puts one bioinformatician between a PI’s sequencing data and the PI’s results is structurally broken in ways that do not get better by working harder within it.
The gap is getting wider. Bioinformatician salaries now average around 80,000 USD for entry-level positions, with master’s-level professionals earning closer to 100,000 USD annually, and the Bureau of Labor Statistics projects 15 percent growth in demand through 2032. Academic labs competing against industry for the same talent pool are not winning that competition. The bioinformatician you interviewed three months ago took a pharma job at 30 percent more than you could offer.
Self-service RNA-seq analysis is not a workaround for this situation. It is the correct architectural response to it.
I made a guide to help wet lab biologists learn computational biology because so many people asked how to get started. The number one question was not about tools or programming languages. It was: where do I even begin?
Why Hiring a Bioinformatician Is Getting Harder
The demand for bioinformatics expertise has grown faster than the pipeline producing trained bioinformaticians. Every major sequencing technology generates more data than its predecessor, and the analytical complexity has grown alongside the data volume. Labs that five years ago sequenced 10 samples per year now sequence 50, and the analysis required per sample has expanded from basic DEG lists to integrated multi-omic analyses.
At the same time, the most capable bioinformaticians have choices. Industry compensation in biotech and pharma consistently exceeds academic salaries by 30 to 50 percent. Data science roles outside biology pay comparably to bioinformatics but require less domain expertise. The talent that might once have taken a postdoc or staff scientist position in an academic lab is increasingly making different calculations.
The result is a hiring market where a wet-lab PI competing for a good bioinformatician is competing against Genentech, Illumina, and Microsoft Research. For most academic groups, that competition is not winnable on compensation. The structural answer is not to compete harder. It is to reduce the dependency.
The Four Analyses Any Wet-Lab Can Run Safely
Not all RNA-seq analysis requires deep bioinformatics expertise. Some of it is well-understood, validated, and directly automatable. The following four categories can be safely run by a trained biologist using a platform with validated defaults, without a bioinformatician actively involved in the execution.
Standard quality control. FastQC report interpretation, MultiQC summary review, adapter trimming assessment, and mapping rate checks all follow documented criteria with clear thresholds. A researcher who knows what a healthy versus unhealthy FastQC report looks like can make the go/no-go decision for each sample. This knowledge is learnable in a half-day and does not require programming.
Alignment and quantification on well-annotated organisms. Running Salmon pseudoalignment against a GENCODE or Ensembl transcriptome for human or mouse is sufficiently validated that automated execution with fixed defaults is appropriate. The tool is deterministic, the reference is well-tested, and the failure modes are documented.
Differential expression on simple designs. A two-condition comparison with three or more replicates per group, no batch effects, and a design formula of ~ condition is the canonical DESeq2 use case. The statistical approach is mature, the defaults are sensible, and the output is interpretable. A researcher who understands what padj and log2FC mean can assess whether the result makes biological sense.
Basic gene ontology enrichment on a finalized DEG list. ORA with a correct background, GSEA with the full Wald statistic ranking, and Benjamini-Hochberg correction applied by default: these are well-defined steps with documented best practices. A researcher who can describe the biological context of their experiment can evaluate whether the enriched terms are plausible.
The Four That Still Need a Human
The categories above are the 90 percent. The following are the 10 percent where computational expertise is not optional.
Custom or complex design matrices. Any experiment with more than two conditions, paired samples, interaction terms, or covariates beyond a simple batch label requires someone who understands the linear model well enough to specify it correctly. A wrong design formula in DESeq2 does not produce an error; it produces quietly wrong results. This is exactly the kind of silent failure that requires expertise to detect.
Batch effect interpretation and correction. Deciding whether a PCA separation is a batch effect or a biological signal, determining whether batch is confounded with condition, and choosing between design-formula modeling and ComBat-Seq correction: these decisions have scientific consequences and require someone who understands what they are doing.
Novel or non-standard methods. Any analysis that deviates from the standard pathway (deconvolution, isoform analysis, multi-omic integration, non-model organism pipelines with custom annotation) requires domain expertise. Platforms cover what they were designed to cover.
Results interpretation. This is the category that is most consistently underestimated. Running the pipeline and interpreting its output are different skills. A DEG list is not a conclusion. Whether the enriched pathways make sense given what you know about the biology, whether the direction of the fold changes is consistent with your experimental perturbation, whether the volcano plot shape looks typical or suspicious: these require biological judgment applied to computational output, and they require someone who has both.
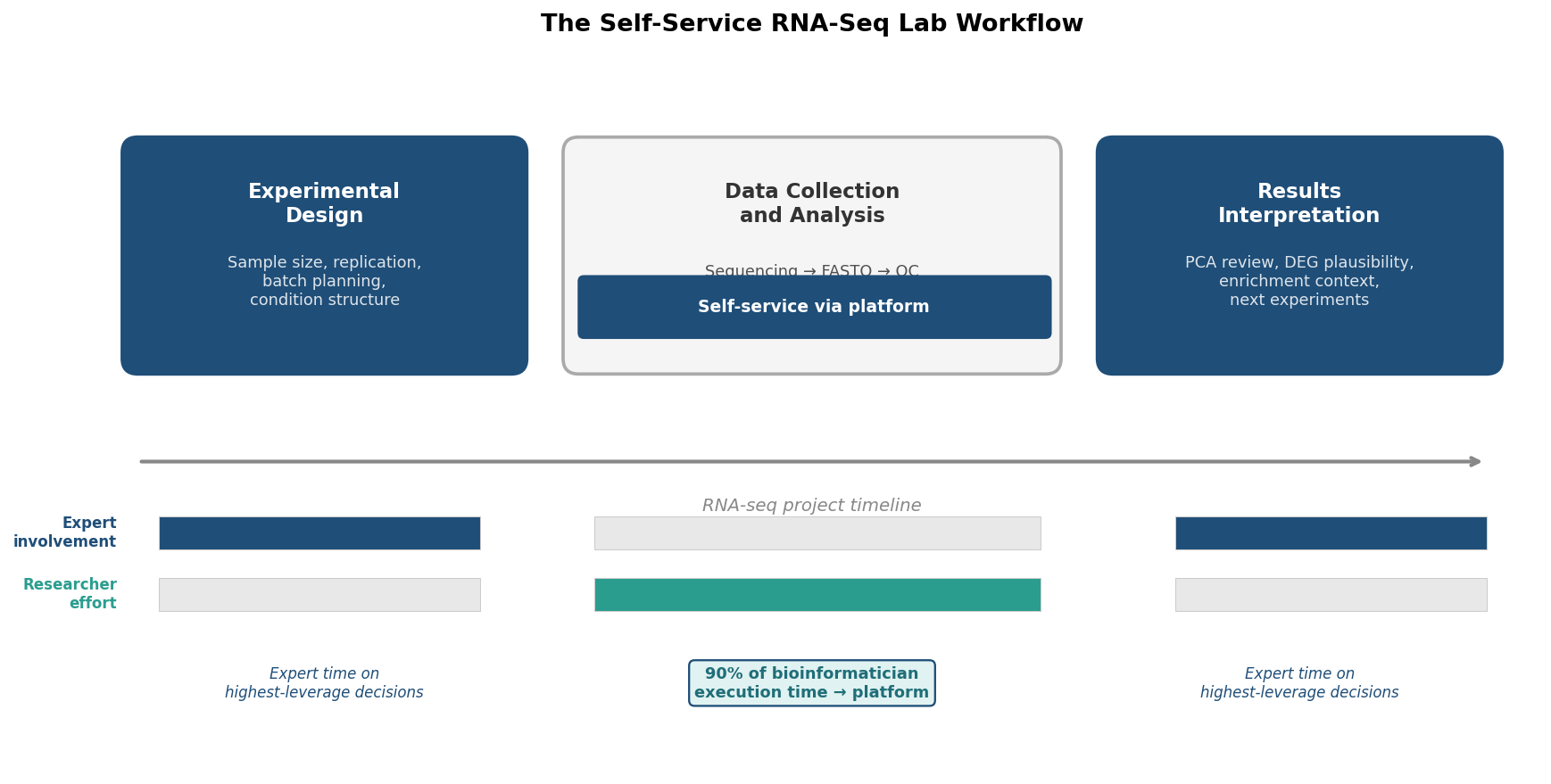
The handoff rule: execution vs interpretation
A useful rule of thumb for dividing work between self-service and expert involvement: the execution of a well-specified analysis can be self-service. The interpretation of results cannot. A wet-lab researcher can run the DESeq2 analysis and produce the DEG list. The judgment call about whether the top hits make biological sense, whether the PCA shows expected patterns, and what follow-up experiments the data suggests still requires someone with the combined biological and statistical context to evaluate it honestly.
The Hybrid Workflow in Practice
The most effective arrangement for a wet-lab group without dedicated bioinformatics support is a hybrid: self-service execution using a platform with validated defaults, combined with periodic expert review at the interpretation stage.
This is not a diminished version of having a dedicated bioinformatician. It is a different allocation of expertise. The bioinformatician’s time is concentrated where it is most valuable, which is at the interpretation stage and at the design stage before sequencing begins. The execution stage, which is typically where most of their time actually goes, is handled by the platform.
A practical implementation for a lab running six to twelve RNA-seq experiments per year:
Before sequencing: consult with a bioinformatician or biostatistician on experimental design. This is the highest-leverage intervention. A confounded design cannot be recovered computationally. This consultation takes two to four hours and should happen before any samples are sent for sequencing.
After sequencing: run quality control and differential expression through a platform. This generates the DEG list, the enrichment summary, the PCA plot, and the methods text. A trained researcher can complete this step in an afternoon for a standard experiment.
At interpretation: bring the results to a bioinformatician or a lab meeting with appropriate expertise. Review the PCA for unexpected separation, discuss whether the top DEGs and enriched pathways are consistent with the biology, and identify follow-up questions. This takes one to two hours per dataset and is the step that determines whether the analysis produces actionable science.
The Cost Comparison
The business case for self-service is clearest when you compare the actual cost of alternatives.
| Analysis path | Typical cost | Turnaround | Reproducible? | Scales with experiments? |
|---|---|---|---|---|
| In-house bioinformatician (full-time) | 80,000 to 120,000 USD/yr salary + benefits | Days to weeks depending on queue | Depends on their practices | Yes, up to their capacity |
| Bioinformatics consulting (per project) | 2,000 to 8,000 USD per standard experiment | 2 to 6 weeks | Depends on consultant | Yes, at per-project cost |
| Core facility analysis | 500 to 2,000 USD per project | 4 to 8 weeks | Often limited | Yes, up to core capacity |
| Self-service platform | Subscription or per-run, typically under 200 USD per experiment | Hours to 1 day | Built in by default | Yes, unlimited |
The self-service platform does not replace the expert consultation at the design and interpretation stages. Those consultations still cost time and sometimes money. What it replaces is the execution stage, which is the stage that takes the most calendar time and consumes the most of a bioinformatician’s or consultant’s hours for work that is, frankly, routine.
Starting With NotchBio
NotchBio is built specifically for this workflow. A wet-lab researcher uploads a sample metadata table and FASTQ paths, selects their organism and annotation version, and gets back quality control reports, differential expression results, enrichment analysis, a PCA plot, a volcano plot, a heatmap, and a complete methods paragraph with tool versions and parameter citations. The run takes under two hours for a standard six-to-twelve sample experiment. The results are ready for the interpretation meeting.
Every run produces a permalink with locked parameters. If a reviewer asks what version of DESeq2 was used, the answer is one link. If you want to rerun the analysis on a revised sample set, you clone the run and change the sample table. Nothing about the pipeline needs to be debugged, reconfigured, or maintained.
Labs that sequence fewer than twenty samples per year and do not have dedicated bioinformatics staff are the labs NotchBio was built for. If that describes your lab, the fastest way to evaluate whether it fits your workflow is to start with a free run at notchbio.app. The first run includes full QC and differential expression on up to twelve samples with no setup required.
The FASTQ files have been on your server for three weeks. They do not have to wait another month.
Related Reading
Further reading
Read another related post
Download RNA-Seq Data from GEO and SRA with sra-tools
Download bulk RNA-seq FASTQ files from GEO and SRA: prefetch, fasterq-dump, pysradb metadata, batch downloads, and fixes for the most common errors.
TutorialHow to Make Volcano and MA Plots in R with ggplot2
Publication-quality volcano and MA plots from DESeq2 results in R: ggplot2 from scratch, ggrepel gene labels, EnhancedVolcano, and how to read them.
TutorialPCA and Clustering for RNA-Seq QC in Python
Python tutorial: normalize RNA-seq counts, run PCA with scikit-learn, build a sample distance heatmap, and spot outliers before differential expression.