From Wet Lab to Dry Lab: A Realistic Map of What to Learn First
Most wet-lab biologists who decide to learn bioinformatics make the same mistake: they try to learn everything at once. They open a Coursera syllabus, see twelve modules spanning Unix, Python, R, statistics, machine learning, and genome assembly, and either freeze or spend six months on material they will never use for RNA-seq analysis. The field does not help. Its learning resources are designed by people who already know what matters, and they rarely say what you can safely ignore.
This post is a map, not a curriculum. It is written for the biologist who has sequencing data to analyze, a PI who expects results, and a finite number of hours per week to spend learning new things. It answers three questions: what do you actually need to learn, in what order, and what can you skip or outsource while you build toward self-sufficiency.
The Three Skills That Actually Matter
For bulk RNA-seq analysis specifically, the skill set that covers 90 percent of what you will do day to day comes down to three areas. Not twelve. Three.
Unix command line basics. Your sequencing files arrive as compressed FASTQ files. Quality control tools, aligners, and quantifiers are all command-line programs. You cannot move, decompress, or run these files without knowing how to navigate a terminal, write a basic loop, and redirect output to a file. You do not need to be a bash programmer. You need to be comfortable enough to not panic when you see a dollar sign prompt.
The specific skills that matter: changing directories, listing files, moving and copying files, decompressing .gz archives with zcat or gunzip, running a command with arguments, and understanding what standard output and standard error mean. That is genuinely it for getting through QC and alignment. Everything more advanced than this you will pick up as you need it.
R for data analysis. The dominant tools for differential expression analysis (DESeq2, edgeR, limma-voom) are R packages. The dominant tools for enrichment analysis (clusterProfiler, fgsea) are R packages. The plotting ecosystem in R (ggplot2) produces the figures that end up in papers. You cannot realistically do RNA-seq analysis in 2025 without R.
You do not need to love R. You do not need to become an R programmer who writes packages. You need to be able to load data from a file, run a function with arguments, understand what a data frame is, and make a basic plot. A motivated biologist can reach functional competence in R within four to six weeks of consistent practice, where consistent means roughly an hour per day.
Statistics for genomics. This is the one that gets skipped most often and causes the most damage. Understanding what a p-value means, why adjusted p-values exist, what a false discovery rate is, what fold change represents on a log scale, and why you need biological replicates to do any of this reliably: these are not advanced topics. They are the minimum context for interpreting your own results without misleading yourself or your PI.
The good news is that you do not need a statistics degree. You need a working understanding of hypothesis testing as applied to count data, and the conceptual difference between statistical significance and biological significance. Two to three weeks of focused reading, combined with running your first DESeq2 analysis while paying attention to the output, will get you there.
What You Can Skip
Knowing what not to spend time on is as important as knowing what to prioritize. The following are real topics that appear in bioinformatics curricula and are genuinely not necessary for RNA-seq analysis in a standard wet-lab research context.
Algorithm internals for aligners. You do not need to understand how the BWT-FM index in STAR works, how suffix arrays are constructed, or the mathematical basis of quasi-mapping in Salmon. You need to know what parameters to set and what the output means. The implementation is someone else’s problem.
Machine learning and deep learning. Unless your project specifically requires it, ML is a distraction at this stage. Feature engineering, neural network architectures, gradient descent: none of this will help you get from FASTQ to a DEG list faster or more accurately. The field has a tendency to conflate “doing bioinformatics” with “applying ML to everything,” and that conflation misleads beginners badly.
Building your own pipeline infrastructure. Snakemake, Nextflow, and Cromwell are excellent tools for scaling and reproducibility. You will eventually want to learn one of them. But spending three weeks learning Nextflow syntax before you have run a single alignment is backwards. Learn the steps first, automate later.
Genome assembly and variant calling. These are entirely separate skill sets from RNA-seq analysis. The tools, the statistics, and the biological questions are different. If your project involves both RNA-seq and variant calling, that is two separate learning tracks. Conflating them adds confusion without adding capability.
The Order That Works
Learning is path-dependent. Some things are much easier to understand once you have seen a related concept in practice. The following sequence reflects what actually makes sense to learn in order, based on what depends on what.
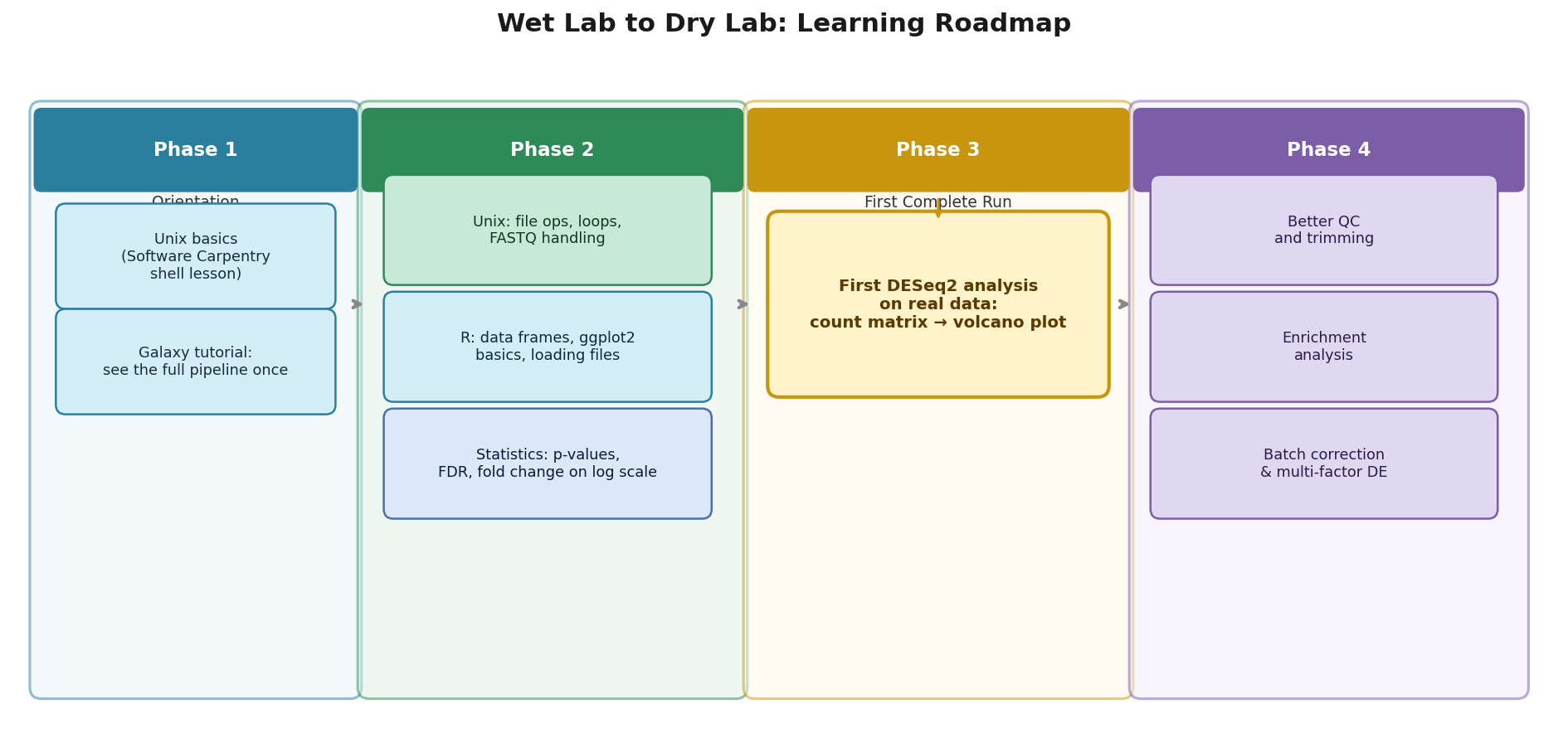
Start with Unix, but stop early. Two weeks of focused Unix practice (Software Carpentry’s shell lesson is the best free starting point) gets you to the level you need. Resist the urge to go deeper before you have a reason.
Next, run your first complete analysis end to end using a tutorial dataset before you touch your real data. Galaxy, the web-based bioinformatics platform, lets you do this without installing anything. The Galaxy Training Network has an RNA-seq tutorial that takes you from FASTQ to differential expression through a browser. Do it once. The goal is not to understand everything; it is to see the whole pipeline as a connected sequence of steps before you learn each step in detail.
Then learn R, but learn it through your analysis, not in the abstract. The mistake is spending six weeks on R syntax exercises before you have data to analyze. The better path is to start with a real count matrix from your Galaxy run and work through loading it, running DESeq2, and interpreting the output. The statistics knowledge you need will surface naturally as you try to understand what DESeq2 is telling you.
By the time you have run differential expression once in R and understood the output table, you have the foundation to go deeper in any direction: better QC, better experimental design, enrichment analysis, batch correction. Everything builds on that first complete pass.
Understand the count matrix before learning DESeq2 internals
Before you read the DESeq2 paper or try to understand negative binomial dispersion estimation, make sure you can answer these three questions about a count matrix: what does each row represent, what does each column represent, and why do two samples from the same condition have different total counts. If you cannot answer those from first principles, the DESeq2 internals will not make sense anyway.
Free Resources That Actually Work
The bioinformatics community is generous with educational materials. The following are the resources that come up repeatedly in community recommendations and that have stood up to years of use by beginners.
The Software Carpentry shell lesson covers Unix basics in a structured, beginner-friendly format with exercises. It is free, browser-accessible, and takes about six hours to complete at a pace that leaves time to practice. It consistently appears at the top of “where should I start” recommendations.
The Bioconductor RNA-seq workflow, available on the Bioconductor website, walks through a complete analysis in R from count matrix to visualization using real data. It is written by the authors of the tools it uses, which means the reasoning behind each step is explained correctly.
The Galaxy Training Network’s RNA-seq tutorial is the best no-installation option for seeing the full pipeline before you commit to learning the command-line tools. It is particularly useful for wet-lab biologists who want to understand the pipeline conceptually without immediately confronting the Unix environment.
R for Data Science by Hadley Wickham, available free online at r4ds.had.co.nz, is the most used introductory R resource in the data science community. It does not focus on bioinformatics specifically, but the R skills it builds transfer directly.
StatQuest with Josh Starmer on YouTube covers RNA-seq statistics in a way that is genuinely accessible to biologists without a statistics background. The DESeq2 video series in particular is worth watching before you run your first analysis, not as a replacement for reading the documentation, but as a conceptual primer.
The Learning Path in One View
The Skill Priority Matrix
The following table summarises which skills to prioritise, which to defer, and which to outsource during each phase of the transition. Use it as a planning tool, not a rigid schedule.
| Skill | Priority | When to learn | Can outsource early? |
|---|---|---|---|
| Unix basics (navigate, copy, decompress) | Essential | Week 1 to 2 | No |
| R basics (load data, run functions, plot) | Essential | Week 3 to 8 | No |
| Statistics (p-value, FDR, fold change) | Essential | Weeks 4 to 8 alongside R | No |
| FastQC and MultiQC interpretation | High | Week 2 to 4 | Yes, short-term |
| DESeq2 or edgeR differential expression | High | Week 6 to 10 | Yes, short-term |
| Enrichment analysis (ORA, GSEA) | Medium | After first DE run | Yes, initially |
| Bash scripting beyond basics | Medium | Month 3 onward | Yes |
| Pipeline managers (Nextflow, Snakemake) | Deferred | Month 6 or later | Yes, long-term |
| Aligner internals (STAR, Salmon mechanics) | Skip | Not required | Yes, indefinitely |
| Machine learning for genomics | Skip | Only if project requires | Yes, indefinitely |
When To Use a Platform vs When To Write the Script
There is a version of the wet-lab-to-dry-lab transition that skips the learning entirely and outsources all analysis to a platform. There is a version that insists on writing every line of code from scratch from day one. Neither extreme serves a working scientist well.
The practical answer is that you need to understand what each step of the pipeline does, even if you are not the one running it. A platform that handles QC, alignment, quantification, and differential expression automatically is not a shortcut that bypasses understanding; it is a tool that produces output you still need to interpret. The biologist who cannot read a volcano plot cannot make good decisions from one, regardless of how it was generated.
The better framing is: outsource the execution while you build the understanding. Running your analysis on a platform while you are learning gives you real results to study and interpret, which is the fastest way to build the statistical intuition you actually need. Once you understand what the pipeline is doing and why each parameter matters, you can decide whether writing your own scripts adds value to your specific workflow.
You will eventually learn Snakemake. You do not need to learn it this week to get publication-quality differential expression results. NotchBio is built for exactly this phase of the transition: it runs QC, alignment, quantification, and differential expression with sensible defaults, surfaces the results in plain language, and generates the methods text your paper will need. It gives you real output to study while you build toward the point where you want to control every parameter yourself.
The goal of the transition is not to become a software engineer who happens to work on biology. It is to become a biologist who can ask precise computational questions about your data and trust the answers you get back. That competence takes time to build regardless of which tools you use along the way.
Related Reading
Further reading
Read another related post
Why Reproducibility Should Not Be Optional in RNA-Seq Pipelines
Run snapshots, version pinning, and locked parameters should be the default, not a feature. A practitioner case for reproducibility-first RNA-seq platforms.
BioinformaticsBulk RNA-Seq for Bacteria: Operons and Why nf-core Breaks
Most bulk RNA-seq pipelines fail silently on bacterial data. Here is what changes for operons, GTF feature mismatches, and DE analysis in prokaryotes.
TutorialPublication-Ready RNA-Seq Plots in ggplot2
Reviewer-ready RNA-seq plots in R: volcano with gene labels, z-score heatmap with annotation bars, PCA with variance explained, and journal export settings.