Bulk RNA-Seq for Bacteria: Operons and Why nf-core Breaks
Run nf-core/rnaseq on a bacterial dataset and watch it fail. Not loudly, with a clear error message explaining what went wrong. Quietly, at the alignment or quantification step, because the GTF file for your organism does not contain the exon feature that the pipeline expects as a hardcoded assumption. The nf-core developers documented this in issue #1512 and resolved it by recommending that users edit the GTF to add synthetic exon features mirroring the CDS coordinates. That workaround tells you everything you need to know about how the standard RNA-seq toolchain was built: with eukaryotes in mind, from the ground up.
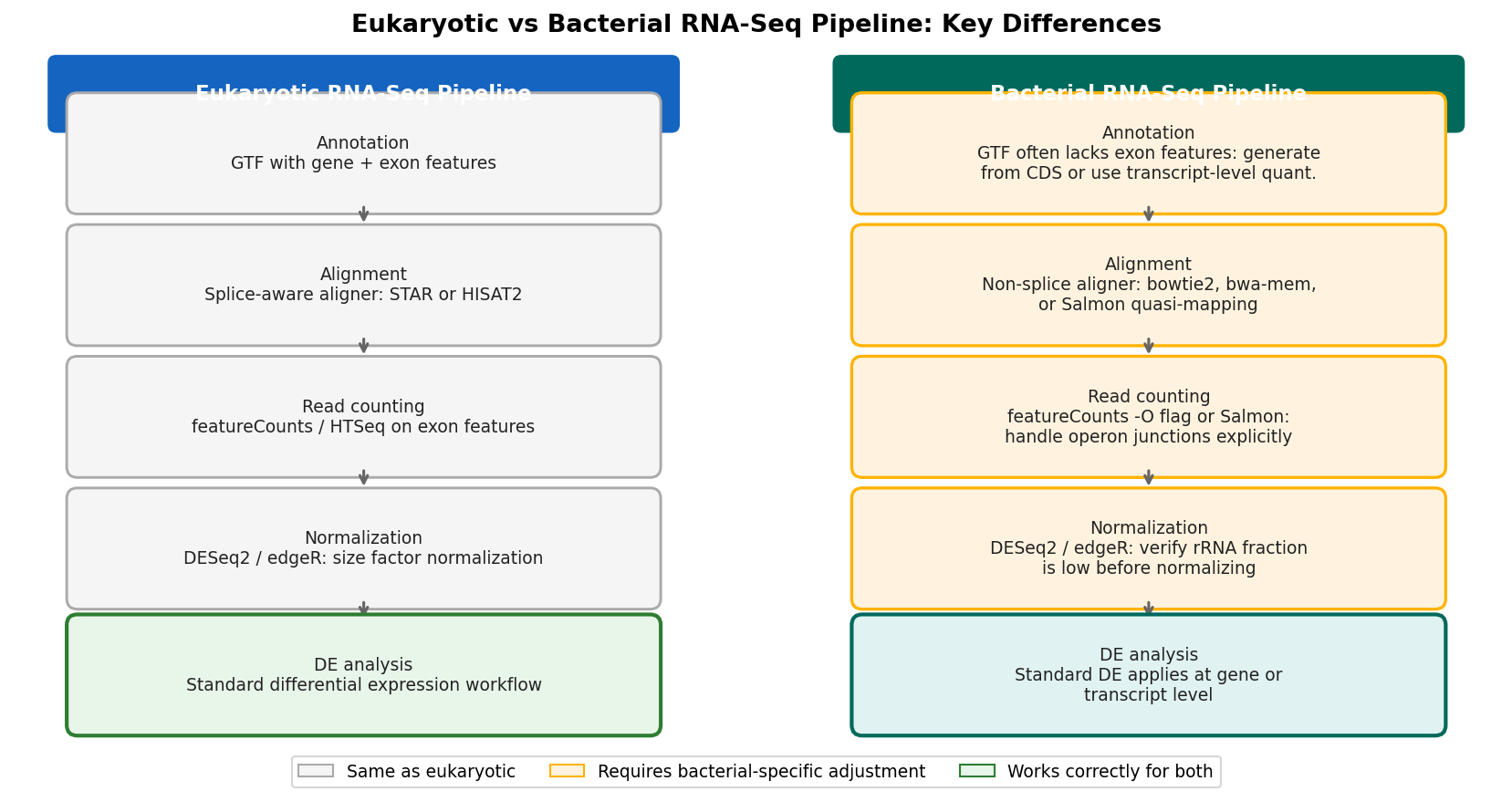
Bacterial transcriptomics is not a minor variation on eukaryotic RNA-seq. The biology is different in ways that ripple through every stage of the pipeline, from annotation file structure through read counting through differential expression modeling. If you are running RNA-seq on a bacterial organism and using a standard eukaryotic pipeline without modification, your results are likely wrong in ways you cannot detect by looking at the output.
This post covers what actually changes and what to do about it.
Why Standard Pipelines Silently Break on Prokaryotes
The root cause is a structural assumption baked into almost every widely used RNA-seq tool: that the genome is organized into genes containing exons, with introns between them. In eukaryotes, pre-mRNA splicing means that reads can span exon-intron boundaries, and aligners like STAR and HISAT2 are specifically designed to handle splice junctions. Annotation files in GTF format reflect this by describing genes as parent features containing exon child features, each with genomic coordinates.
Bacterial genomes do not have introns. Bacterial genes are contiguous. A GTF or GFF3 file for a well-annotated bacterial genome will typically contain gene features and CDS features, but not exon features, because there are no exons to annotate. When featureCounts, HTSeq, or the nf-core pipeline’s annotation parsing step looks for exon features to count reads against, it finds nothing and either raises an error or, more dangerously, produces a count matrix of zeros without explaining why.
The STAR aligner handles bacterial data incorrectly in a second way. Its splice-aware alignment algorithm will attempt to find splice junctions that do not exist, sometimes misaligning reads that cross gene boundaries in an operon. The correct approach for bacteria is to use an aligner that does not assume splicing, either by disabling splice-aware mode in STAR or by using a tool like bowtie2 or bwa-mem that does not attempt junction detection at all.
Standard pipelines fail silently on bacterial GTF files
If your bacterial GTF or GFF3 file lacks exon-level features, tools like featureCounts and HTSeq will either error out or return an all-zero count matrix without a clear explanation. Check your annotation file for the presence of exon features before running any counting step. If they are absent, you need to either generate them from CDS coordinates or use a counting approach that works directly from CDS or gene features.
Operons: The Feature That Changes Everything
The defining organizational feature of bacterial transcription is the operon: a cluster of functionally related genes transcribed as a single polycistronic mRNA under the control of a shared promoter. A read sequenced from a polycistronic transcript can span the boundary between two adjacent genes within the same operon. In eukaryotes, a read that spans two genes would be evidence of a chimeric or artifactual sequence. In bacteria, it is normal biology.
This creates an immediate problem for read counting. Standard counting tools assign a read to a feature based on where it maps in the genome. A read that maps to the junction between mcrB and mcrC within the McrBC operon in E. coli is genuinely derived from both genes simultaneously. The counting tool must decide: does this read count toward mcrB, toward mcrC, toward both, or toward neither?
The default behavior of most counting tools is to mark such reads as ambiguous and exclude them from the count for any gene. For a bacterial dataset where a substantial fraction of reads may span operon junctions, this means systematically undercounting transcription from genes that are part of multi-gene operons. The degree of undercounting is not random. It is correlated with operon membership, which is itself correlated with biological function. The bias is not noise; it is structured.
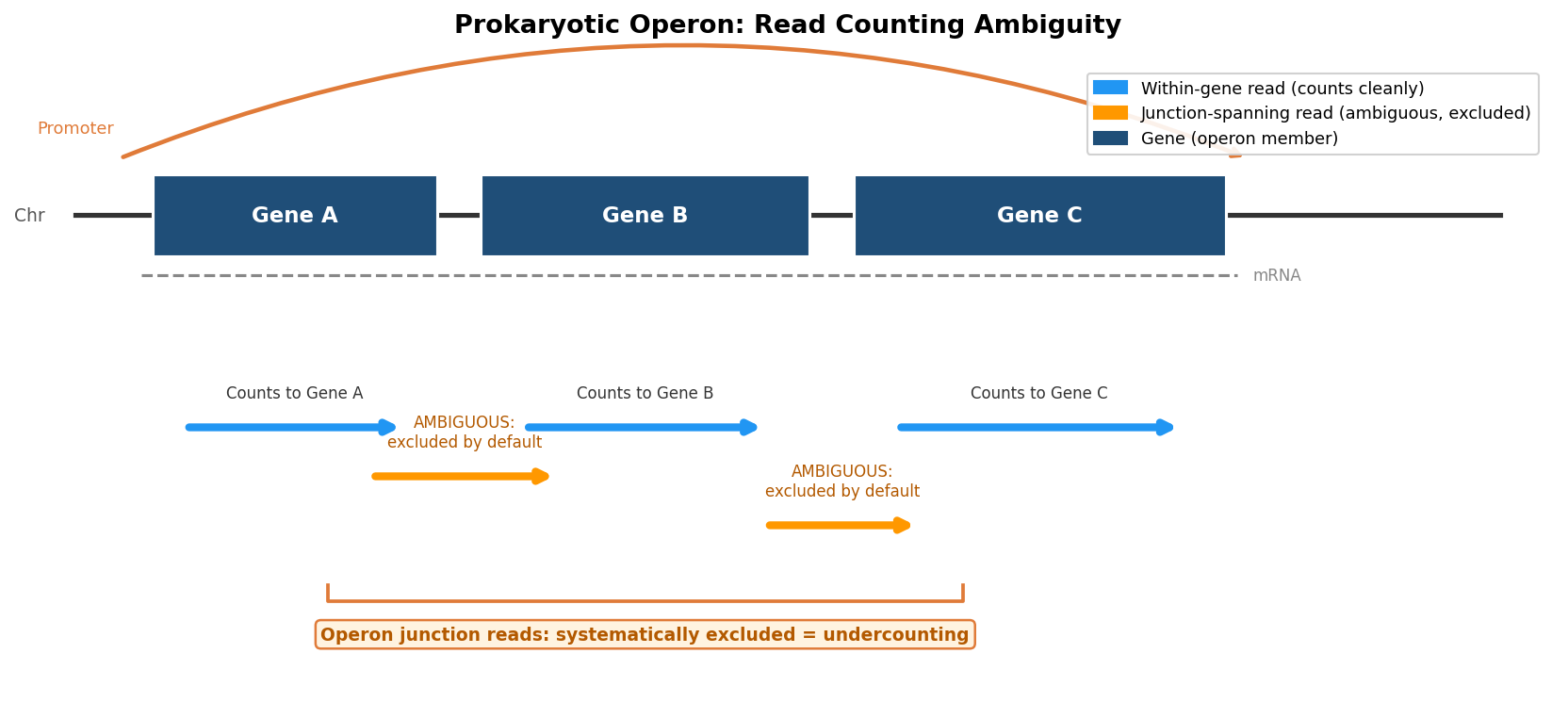
Approaches to Operon Read Counting
Several strategies exist for handling operon junction reads, each with different tradeoffs.
Allow multi-feature counting. featureCounts can be run with the -O flag, which allows reads that overlap multiple features to be counted toward each overlapping feature rather than being marked ambiguous. For operon junction reads, this means the read is counted once toward each gene it overlaps. This inflates the raw counts for genes at operon junctions, but for a dataset where most reads are within-gene rather than spanning junctions, the inflation is modest and the alternative of systematic exclusion is worse. This choice must be documented in the methods section because it affects count matrix interpretation.
Count at the operon level. If your annotation includes curated operon boundaries, counting reads against operons rather than individual genes sidesteps the junction problem entirely. The operon becomes the unit of quantification, and within-operon read assignment ambiguity disappears because the operon is a single contiguous feature. Differential expression then gives you operon-level results, which must be further interpreted to attribute changes to specific genes within the operon. The OperonDB and RegulonDB databases provide operon annotations for well-studied organisms like E. coli and B. subtilis.
Transcript-level quantification with Salmon. The approach favored by experienced practitioners in the community is to build a transcriptome FASTA from the GTF annotation and use Salmon in quasi-mapping mode with a genome decoy. This avoids the splice-aware aligner problem entirely and works correctly from CDS-level features. The decoy genome ensures that reads originating from intergenic regions or non-annotated features do not get spuriously assigned to annotated transcripts.
The Salmon workflow for a bacterial organism looks like this:
# Extract transcript sequences from genome and CDS annotationgffread annotation.gff -g genome.fasta -w transcriptome.fasta
# Build decoy-aware index using genome as decoycat transcriptome.fasta genome.fasta > gentrome.fastagrep "^>" genome.fasta | cut -d " " -f 1 | tr -d ">" > decoys.txtsalmon index -t gentrome.fasta -d decoys.txt -i salmon_index --keepDuplicates
# Quantifysalmon quant -i salmon_index -l A \ -1 sample_R1.fastq.gz -2 sample_R2.fastq.gz \ --validateMappings -o sample_quantThe --keepDuplicates flag matters here. Bacterial genomes often contain paralogous genes with near-identical sequences, and without this flag Salmon may merge what it considers duplicate sequences in ways that lose biologically meaningful distinctions.
Overlapping Genes and Ambiguous Coordinates
Bacterial genomes are densely packed. Genes frequently overlap by a few base pairs at their boundaries, often because stop codons from one gene overlap with the start codon of the next. In some organisms, particularly in compact genomes, genes on opposite strands overlap substantially.
This creates a second source of read assignment ambiguity distinct from the operon problem. A read mapping to a region where two genes overlap on the same strand, or where genes on opposite strands share coordinates, cannot be unambiguously assigned to one gene by coordinate-based counting. The featureCounts behavior for overlapping features depends on the -O and --fracOverlap settings, and the defaults are conservative enough that a significant fraction of reads in a dense bacterial genome may be excluded.
The practical recommendation is to inspect your genome annotation for the extent of gene overlap before choosing a counting strategy. For most well-annotated bacterial reference genomes from NCBI RefSeq, the overlap frequency is manageable and the multi-feature counting flag resolves most cases. For poorly annotated or highly compact genomes, the Salmon transcript-level approach is more robust because it sidesteps coordinate-based overlap entirely.
Differential Expression: What Changes and What Does Not
The good news about differential expression analysis in bacteria is that DESeq2 and edgeR both work correctly on prokaryotic count data from a statistical standpoint. The negative binomial model does not assume anything about the eukaryotic or prokaryotic origin of the counts. If the count matrix is correctly constructed, the DE analysis proceeds normally.
What deserves scrutiny is the composition of the library before normalization.
Bacterial RNA-seq without rRNA depletion contains an enormous fraction of ribosomal RNA, sometimes 90 percent or more of all reads. If your rRNA depletion was incomplete, the size factors that DESeq2 estimates during normalization will be dominated by the rRNA counts rather than by the messenger RNA you actually want to compare. Check your alignment statistics for the fraction of reads mapping to rRNA genes before proceeding. If rRNA contamination is above 20 to 30 percent, the normalization assumptions are likely violated and results require careful interpretation.
Sequencing depth also deserves attention. Bacterial genomes are small compared to eukaryotic genomes, which means you need far fewer reads to achieve adequate coverage. Five to ten million reads per sample is typically sufficient for standard differential expression in bacteria, compared to 20 to 30 million for human RNA-seq. Under-sequencing relative to the rRNA content of your library leaves you with too few mRNA reads to achieve statistical power for lowly expressed genes.
The GTF Fix: Generating Synthetic Exon Features
If you need to use a pipeline that requires exon features and your bacterial annotation does not provide them, the most straightforward fix is to use AGAT or awk to generate synthetic exon features from the CDS coordinates:
# Using AGAT to add exon features derived from CDSagat_convert_sp_gff2gtf.pl --gff annotation.gff -o annotation_with_exons.gtf
# Alternatively, use awk to duplicate CDS records as exon recordsawk 'BEGIN{OFS="\t"} $3=="CDS" {$3="exon"; print}' annotation.gtf \ >> annotation_with_exons.gtfThe awk approach is simpler but does not handle edge cases like CDS features that already have associated exon features. The AGAT approach is more robust but requires installation. Either way, validate the resulting GTF by checking that exon features exist, their coordinates match the source CDS coordinates, and the attribute fields (gene ID, transcript ID) are correctly preserved.
This fix allows standard counting tools to run, but it does not solve the operon junction counting problem. For that, the -O flag or the Salmon transcript approach remains necessary.
NotchBio handles GTF normalization automatically, detecting the absence of exon features and generating them from CDS coordinates before the counting step runs. A full prokaryote mode with operon-aware read counting is on the roadmap. If your lab regularly runs bacterial RNA-seq and wants to discuss early access, you can reach out through the site.
Related Reading
Further reading
Read another related post
What Is a Count Matrix and Why Does It Matter
Raw counts, TPM, FPKM, and DESeq2-normalized values each represent expression differently. What each one is, why it matters, and which to use downstream.
Research GuideExperimental Design Mistakes in Differential Expression
Replicates, confounders, paired designs, and pseudoreplication: the experimental design decisions that decide whether your DESeq2 results hold up.
Research GuideWhy Your Choice of Reference Genome Changes Your Results
GENCODE, Ensembl, UCSC, and RefSeq annotate the same genome differently. How that choice changes RNA-seq alignment, quantification, and your DEG list.