GTF and GFF Files: Why They Hurt and How To Tame Them
You download a GTF file for your organism of interest. You run it through your aligner. The aligner either crashes, silently produces wrong counts, or succeeds in a way that only reveals its problems three steps later when featureCounts cannot find the feature type it was told to look for. You download a second GTF from a different database for the same genome. It is structured differently. You spend an afternoon figuring out why two annotation files for the same genome disagree on where a gene ends.
Every generation of bioinformaticians goes through this. It is not a rite of passage. It is a consequence of annotation formats that were designed incrementally by different groups with different priorities over several decades, and a community norm that treats getting your GTF working as the researcher’s problem rather than an infrastructure problem worth solving properly.
This post will not make GTF files fun. It will give you a map of what you are actually dealing with, the specific failures that appear most often, and the fastest paths through them.
Why GTF and GFF Formats Disagree
GTF and GFF3 are not the same format with different file extensions, though they are often treated as interchangeable. GTF (Gene Transfer Format) emerged from the Ensembl project and the early microarray era as a simpler derivative of GFF2. GFF3 (General Feature Format version 3) is a later, more formally specified standard with richer hierarchical relationships between features. The two formats have different field conventions, different ways of representing parent-child relationships between genes, transcripts, and exons, and different conventions for attribute quoting and key-value separation.
The practical consequence is that a tool expecting GTF format will often fail on a GFF3 file, and vice versa, even when both files describe the same genome annotation. This is predictable and unavoidable given the history.
The source divergence compounds the format divergence. NCBI, Ensembl, and GENCODE all provide annotation files for the human genome, and all three differ in meaningful ways beyond format. NCBI uses its own chromosome naming convention (NC_000001.11 for chromosome 1 in GRCh38). Ensembl uses the bare chromosome number (1). UCSC uses the chr prefix (chr1). Using a STAR index built with one naming convention and a GTF file using a different one produces zero-mapped reads with no error message, which is one of the most confusing failure modes in RNA-seq analysis.
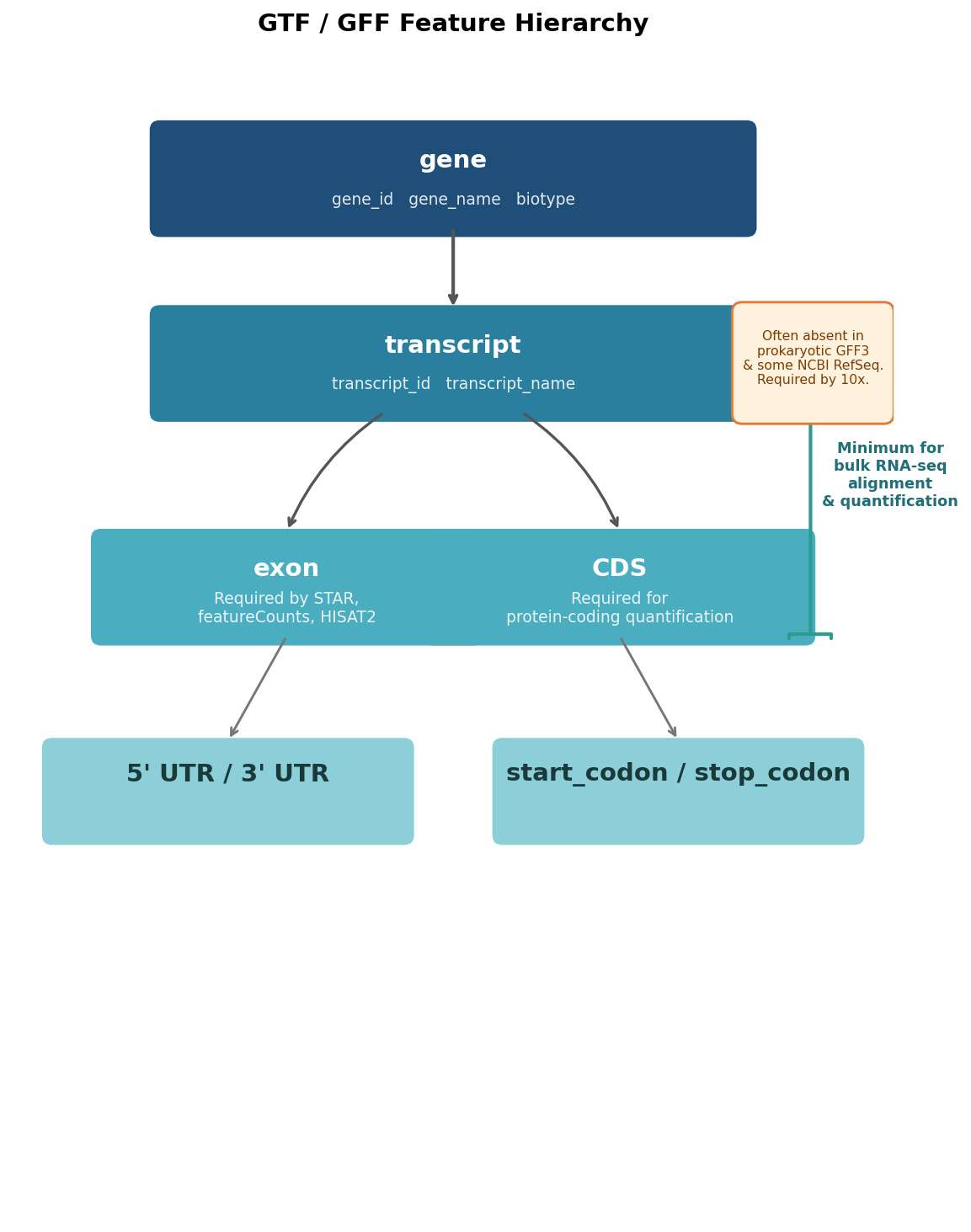
Feature hierarchy conventions also differ. GENCODE GTF files contain gene, transcript, exon, CDS, UTR, and start/stop codon features. NCBI RefSeq GFF3 files structure the same information differently, with mRNA as the parent of exon rather than transcript, and region features at the top level that some tools interpret as chromosome-level entries and others ignore. AGAT, the self-described GTF/GFF Swiss army knife, handles the conversion between these structures but introduces its own complications.
The Specific Gotchas That Break Pipelines
Understanding the general history is useful context. Understanding the specific failure modes is what saves you time at 11pm when your alignment job is failing.
Missing transcript-level features in prokaryotic GFF3 files. Bacterial genome annotations from NCBI RefSeq typically structure genes with CDS features as direct children of gene features, with no intermediate mRNA or transcript entry. Tools like 10x Genomics Cell Ranger and some versions of the nf-core RNA-seq pipeline explicitly require a transcript-level entry between the gene and the exon. When it is missing, the pipeline fails with a message about unrecognised feature types or, worse, silently produces a count matrix with zeros for every gene.
The nf-core/rnaseq GitHub issue tracker has a well-documented thread on this exact problem: the pipeline would fail on prokaryotic GTF files because the assumption of an exon feature was baked into the workflow. The resolution was to edit the GTF to add synthetic transcript entries, which AGAT can do but does not always do correctly on the first attempt.
Chromosome naming mismatches. As described above, this is the silent killer. STAR builds an index using the chromosome names in the FASTA file. If the GTF uses different chromosome names, STAR will report a very high mapping rate against the genome but zero or near-zero reads will be assigned to genes, because the GTF chromosome names do not match the index. There is no error. The numbers just do not make sense.
Check this before alignment: grep "^>" genome.fa | head -5 shows your FASTA chromosome names. cut -f1 annotation.gtf | sort -u | head -5 shows your GTF chromosome names. They must match exactly, including the chr prefix or lack thereof.
Coordinates that extend beyond the chromosome length. Some GFF3 files from NCBI contain feature coordinates that exceed the reported length of the chromosome, usually by a few bases. This is technically a malformed file. Most tools ignore it silently. A few, including some versions of AGAT and some validation tools, will either fail or strip the affected features during processing, causing genes near chromosome ends to disappear from your count matrix.
Attribute quoting differences. GTF format uses double-quoted attribute values: gene_id "ENSG00000139618". GFF3 format uses unquoted values in key=value notation: ID=gene-BRCA2. Tools parsing GTF that receive GFF3 input, or vice versa, may parse attribute fields incorrectly and produce empty or malformed feature names. This is hard to diagnose from error messages alone because the tool often succeeds but assigns all reads to a single gene with a garbled name.
AGAT: Where It Helps and Where It Hurts
AGAT (Another GTF/GFF Analysis Toolkit) is the community’s most-used tool for GTF/GFF manipulation and format conversion. It is genuinely useful, and it is also the source of some of the most confusing secondary problems in GTF troubleshooting.
Where AGAT reliably helps: converting GFF3 to GTF format when the input is well-formed, adding transcript-level entries to files that only have gene and exon, fixing simple coordinate errors, and checking files for consistency against the GFF3 specification. The agat_convert_sp_gff2gtf.pl script handles the most common conversion use case adequately for most Ensembl and GENCODE source files.
Where AGAT introduces problems: NCBI RefSeq GFF3 files contain region features at the top level that represent chromosomes, scaffolds, and organelles. AGAT sometimes treats these as gene-level entries during conversion, which can inflate your annotation with thousands of spurious features. The output GTF may be syntactically valid while being scientifically wrong. Filtering the output with grep -v "^#" annotation.gtf | awk '$3 != "region"' after conversion is a safe habit.
AGAT also performs semantic inference during conversion, meaning it will add features it believes are implied by the structure of the input file. In most cases this is helpful. In cases where the input file has non-standard structure, AGAT may infer the wrong feature hierarchy and produce a GTF that is internally consistent but does not match the original annotation. When in doubt, validate the output with agat_sq_stat_basic.pl and compare gene counts to the source.
Validate AGAT output before alignment
AGAT performs semantic inference during GFF3-to-GTF conversion. Always check the gene and transcript counts in the converted file: agat_sq_stat_basic.pl —gff converted.gtf. If the count differs substantially from the source annotation, the conversion introduced errors. Compare a few specific genes manually using grep before proceeding to alignment.
A Normalization Script That Handles 80 Percent of Cases
For the most common scenario, a well-formed GFF3 or GTF file from Ensembl, GENCODE, or NCBI that needs to be used with a tool expecting standard GTF, the following bash workflow resolves the most frequent issues without AGAT.
#!/bin/bash# gtf_normalize.sh : basic GTF sanity checks and chr-prefix normalization# Usage: bash gtf_normalize.sh input.gtf genome.fa output.gtf
INPUT_GTF=$1GENOME_FA=$2OUTPUT_GTF=$3
# Step 1: Check chromosome naming in genome FASTAecho "=== Genome chromosome names (first 5) ==="grep "^>" "$GENOME_FA" | head -5 | sed 's/>.*//' | cut -d' ' -f1
# Step 2: Check chromosome naming in GTFecho "=== GTF chromosome names (first 5 unique) ==="grep -v "^#" "$INPUT_GTF" | cut -f1 | sort -u | head -5
# Step 3: Detect chr-prefix mismatch and fix if neededGENOME_HAS_CHR=$(grep "^>" "$GENOME_FA" | head -1 | grep -c "^>chr")GTF_HAS_CHR=$(grep -v "^#" "$INPUT_GTF" | head -1 | grep -c "^chr")
if [ "$GENOME_HAS_CHR" -eq 1 ] && [ "$GTF_HAS_CHR" -eq 0 ]; then echo "Adding chr prefix to GTF chromosome names..." grep -v "^#" "$INPUT_GTF" | sed 's/^\([0-9XYM]\)/chr\1/' > "$OUTPUT_GTF"elif [ "$GENOME_HAS_CHR" -eq 0 ] && [ "$GTF_HAS_CHR" -eq 1 ]; then echo "Stripping chr prefix from GTF chromosome names..." grep -v "^#" "$INPUT_GTF" | sed 's/^chr//' > "$OUTPUT_GTF"else echo "Chromosome naming consistent. Copying GTF..." grep -v "^#" "$INPUT_GTF" > "$OUTPUT_GTF"fi
# Step 4: Check for transcript-level features (required by many tools)TRANSCRIPT_COUNT=$(grep -v "^#" "$OUTPUT_GTF" | awk '$3 == "transcript"' | wc -l)echo "=== Transcript feature count: $TRANSCRIPT_COUNT ==="if [ "$TRANSCRIPT_COUNT" -eq 0 ]; then echo "WARNING: No transcript features found. Tools requiring transcript level may fail." echo "Consider running: agat_convert_sp_gff2gtf.pl --gff $INPUT_GTF -o $OUTPUT_GTF"fi
# Step 5: Check for coordinates beyond chromosome bounds (basic)echo "=== Max coordinate in GTF ==="grep -v "^#" "$OUTPUT_GTF" | awk '{if($5>max) max=$5} END{print max}'This script does not replace AGAT for complex conversions. It catches the two most common immediate failures (chr-prefix mismatch and missing transcript features) before you discover them through a failed alignment run.
The Source-by-Source Decision Matrix
| Source | Format | Chr naming | Has transcript level | Common problems | Recommended use |
|---|---|---|---|---|---|
| Ensembl | GTF | No chr prefix (1, 2, X) | Yes | Extra scaffolds and patches inflate annotation | Remove non-primary contigs: grep ”^[0-9XYM]“ |
| GENCODE | GTF | chr prefix (chr1, chrX) | Yes | Very large file, includes pseudogenes and lncRNA by default | Use primary assembly file |
| NCBI RefSeq | GFF3 | NC_ accessions | Usually for eukaryotes | Region features, NC_ naming mismatches, no transcript in prokaryotes | Convert with AGAT, validate output |
| UCSC | GTF | chr prefix (chr1, chrX) | Yes | Based on NCBI or Ensembl with coordinate lift-over; may lag current release | Match genome assembly version carefully |
| Ensembl Bacteria | GFF3 | Accession-based | No (prokaryote) | No exon features in some releases | Add synthetic transcript and exon features |
When To Download a Different Version vs When To Fix What You Have
The most time-efficient decision rule is this: if you are more than one conversion step away from a valid GTF for your tool, download a different source file.
AGAT is reliable for one-step conversions: GFF3 to GTF, adding transcript features to a well-formed file, stripping region entries from a RefSeq file. If you find yourself running AGAT, validating the output, finding problems, running AGAT again with different flags, and still getting unexpected gene counts, the time cost of that iteration almost always exceeds the time cost of finding a pre-converted GTF from a different source.
For human and mouse, Ensembl and GENCODE provide high-quality GTF files that work with STAR, HISAT2, Salmon, and featureCounts with minimal preprocessing. The chr-prefix difference between Ensembl and GENCODE is the only regular mismatch you need to manage, and the normalization script above handles it in seconds.
For less-studied organisms, NCBI RefSeq is often the only option. In those cases, AGAT conversion is necessary, and the validate-before-proceeding habit is mandatory.
For prokaryotes specifically, adding synthetic transcript and exon entries from AGAT is the standard path. Verify the output transcript count matches your expected gene count and inspect three to five genes manually before running alignment.
NotchBio validates uploaded GTF and GFF files automatically before any pipeline run begins, flagging missing transcript-level features, chr-prefix mismatches against the selected reference genome, and coordinates that exceed chromosome bounds. For most standard organisms, it selects and validates the reference annotation without requiring a manual download.
Related Reading
Further reading
Read another related post
Raw Reads to Counts: The Bulk RNA-Seq Pipeline Explained
Every computational step in bulk RNA-seq, explained: from FASTQ quality control through trimming, alignment, and quantification to your final count matrix.
Research GuideWhat Are Batch Effects in RNA-Seq (and Why They Ruin Results)
What batch effects are, why they happen in bulk RNA-seq, and how they quietly corrupt your differential expression results — the concepts to grasp first.
AnnouncementWelcome to the NotchBio Blog
Introducing the NotchBio blog — your source for RNA-seq analysis insights, bioinformatics tutorials, and platform updates.