Salmon From FASTQ to Counts: A Complete Tutorial
Salmon has become the default quantification tool for standard bulk RNA-seq differential expression workflows. It does not produce a BAM file. It does not attempt to find the genomic coordinates of every read. Instead, it asks which transcripts in a reference are consistent with each read, distributes counts probabilistically across compatible transcripts, and reports estimated counts and TPM per transcript in a few minutes per sample. For any analysis whose goal is differential expression against an annotated transcriptome, this is both faster and at least as accurate as alignment-based approaches.
This tutorial covers the complete Salmon workflow end-to-end: building a decoy-aware index, running quantification with the flags that matter, importing results into R with tximport, feeding directly into DESeq2, and checking that the quantification rates tell a sensible story. Every command runs. Every R code block is copy-pasteable.
Before You Start: What You Need
Salmon itself, a reference transcriptome FASTA, and the matching genome FASTA for building the decoy-aware index. For human or mouse, use GENCODE or Ensembl releases: these are internally consistent between the transcriptome sequences and the gene annotation GTF you will need later for the transcript-to-gene mapping.
# Install Salmon (Linux / macOS)# Option 1: conda (recommended for reproducibility)conda install -c bioconda salmon
# Option 2: download pre-compiled binarywget https://github.com/COMBINE-lab/salmon/releases/download/v1.10.3/salmon-1.10.3_linux_x86_64.tar.gztar -xzf salmon-1.10.3_linux_x86_64.tar.gzexport PATH=$PATH:$(pwd)/salmon-latest_linux_x86_64/bin
# Verifysalmon --version # should print salmon 1.10.3 or current release
# Download reference files (human GRCh38, GENCODE release 45)wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_45/gencode.v45.transcripts.fa.gzwget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_45/GRCh38.primary_assembly.genome.fa.gzwget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_45/gencode.v45.annotation.gtf.gzAlways use a decoy-aware index: it takes 10 extra minutes and improves accuracy
A standard Salmon index is built from transcriptome sequences alone. A decoy-aware index also includes the genome sequence as a decoy, allowing Salmon to identify reads that map to genomic regions outside annotated transcripts and exclude them from quantification rather than spuriously assigning them to a transcript. For human data, the decoy-aware index consistently reduces mapping-rate inflation and produces more accurate abundance estimates, particularly for genes near pseudogenes or repetitive regions. The index build takes roughly 10 extra minutes and 20 extra GB of disk. It is worth it.
Step 1: Build the Decoy-Aware Index
The decoy-aware index construction concatenates the transcriptome FASTA with the genome FASTA and records the genome chromosome names as decoys. Salmon then uses the genome sequences to catch reads that should not be assigned to any transcript.
# Step 1a: Build the gentrome (transcriptome + genome combined FASTA)cat gencode.v45.transcripts.fa.gz GRCh38.primary_assembly.genome.fa.gz \ > gentrome.fa.gz
# Step 1b: Extract chromosome names from genome FASTA to serve as decoy listgrep "^>" <(zcat GRCh38.primary_assembly.genome.fa.gz) \ | cut -d " " -f 1 \ | tr -d ">" \ > decoys.txt
# Verify: decoys.txt should contain chromosome names like chr1, chr2, ..., chrX, chrY, chrMhead -5 decoys.txt
# Step 1c: Build the index# --threads 8: parallelise; adjust to your available cores# --gencode: strips version numbers from GENCODE transcript IDs (e.g., ENST00000456328.2 -> ENST00000456328)# required for tximport tx2gene mapping to work correctlysalmon index \ --transcripts gentrome.fa.gz \ --decoys decoys.txt \ --index salmon_index_grch38_gencode45 \ --gencode \ --threads 8
# Index build takes 30-50 minutes for the human genome, ~20 GB diskThe --gencode flag strips version suffixes from transcript IDs. Without it, the transcript ID in the Salmon output (ENST00000456328.2) will not match the transcript IDs in the tx2gene mapping table (ENST00000456328), causing tximport to fail silently with many unmatched transcripts.
Step 2: Quantify Each Sample
Run salmon quant on each pair of FASTQ files. The key flags are explained inline:
# Quantify one samplesalmon quant \ --index salmon_index_grch38_gencode45 \ --libType A \ --mates1 sample_R1.fastq.gz \ --mates2 sample_R2.fastq.gz \ --validateMappings \ --gcBias \ --seqBias \ --numBootstraps 30 \ --threads 8 \ --output salmon_output/sample_01
# For a batch of samples with bash loopfor SAMPLE in sample_01 sample_02 sample_03 sample_04 sample_05 sample_06; do salmon quant \ --index salmon_index_grch38_gencode45 \ --libType A \ --mates1 fastq/${SAMPLE}_R1.fastq.gz \ --mates2 fastq/${SAMPLE}_R2.fastq.gz \ --validateMappings \ --gcBias \ --seqBias \ --numBootstraps 30 \ --threads 8 \ --output salmon_output/${SAMPLE}done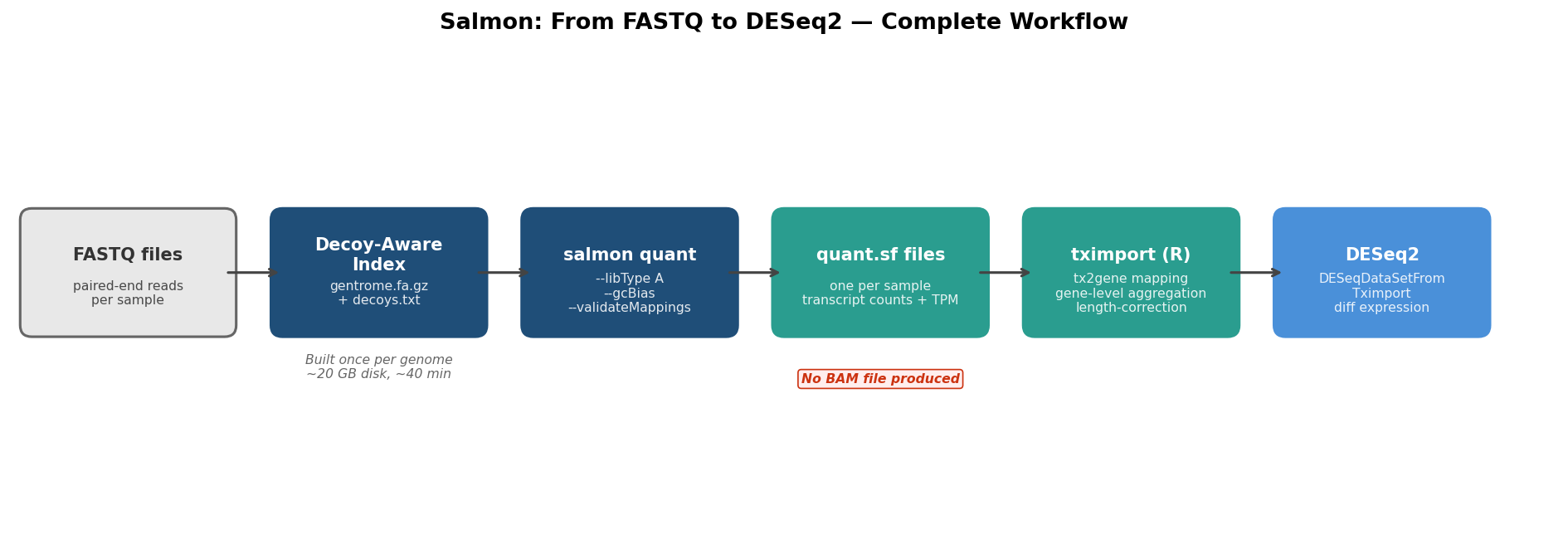
The parameters that matter for standard RNA-seq:
| Flag | What it does | When to use |
|---|---|---|
--libType A | Auto-detect library strandedness | Always; removes need to specify strand manually |
--validateMappings | Run quasi-mapping validation pass for higher accuracy | Always; small speed cost, meaningfully better results |
--gcBias | Correct for GC content bias in fragment generation | Always recommended for Illumina data |
--seqBias | Correct for sequence-specific biases at read ends | Always recommended; minimal cost |
--numBootstraps 30 | Bootstrap resampling for uncertainty estimation | Required if using sleuth or swish for DE; optional for DESeq2 |
--keepDuplicates | Do not discard duplicate sequences during indexing | Use for organisms with paralogous gene families; default OFF |
--threads N | Parallel processing threads | Set to available cores; 8-16 is typical |
--rangeFactorizationBins 4 | Improves accuracy for reads mapping to many positions | Useful for highly repetitive transcriptomes |
Step 3: QC Checks on Mapping Rates
After quantification, check the mapping rate for every sample before proceeding. A healthy Salmon mapping rate for a standard poly(A)-selected human RNA-seq library against a GENCODE transcriptome is typically between 80 and 95 percent. Rates below 70 percent are a warning sign worth investigating.
# Extract mapping rates from all samplesfor SAMPLE in $(ls salmon_output/); do RATE=$(grep "Mapping rate" salmon_output/${SAMPLE}/logs/salmon_quant.log \ | awk '{print $NF}') echo "${SAMPLE}: ${RATE}"done# Parse mapping rates in R for plottingmapping_rates <- data.frame( sample = c("sample_01", "sample_02", "sample_03", "sample_04", "sample_05", "sample_06"), rate = c(91.2, 90.8, 88.4, 92.1, 89.7, 91.5) # replace with actual values)
# Flag samples below thresholdthreshold <- 80mapping_rates$flag <- ifelse(mapping_rates$rate < threshold, "LOW", "OK")
cat("Samples with mapping rate below", threshold, "%:\n")print(mapping_rates[mapping_rates$flag == "LOW", ])Low mapping rates have three common causes: high rRNA contamination (the reads map to the genome but not to annotated mRNA transcripts), FASTQ files from a different organism than the index, or a transcriptome-genome version mismatch (GENCODE release 45 transcriptome paired with a GRCh38.p13 genome rather than GRCh38.p14). The last cause is particularly insidious because it often gives a plausible-looking mapping rate slightly below expectations rather than a catastrophic failure.
Step 4: Build the tx2gene Mapping Table
tximport aggregates transcript-level Salmon output to gene-level counts. It needs a two-column mapping from transcript IDs to gene IDs. The cleanest way to build this from GENCODE annotation is with the txdbmaker or tximeta package:
# Option 1: Build tx2gene from the GTF with txdbmaker (Bioconductor)if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")BiocManager::install("txdbmaker")
library(txdbmaker)
txdb <- txdbmaker::makeTxDbFromGFF( "gencode.v45.annotation.gtf.gz", format = "gtf")
k <- keys(txdb, keytype = "TXNAME")tx2gene <- select(txdb, keys = k, columns = c("TXNAME", "GENEID"), keytype = "TXNAME")
# tx2gene should have two columns: TXNAME (transcript ID) and GENEID (gene ID)head(tx2gene)# Option 2: Extract directly from GTF with a simple parse (faster, no TxDb)library(dplyr)
gtf_lines <- readLines(gzcon(file("gencode.v45.annotation.gtf.gz", "rb")))tx_lines <- gtf_lines[grepl('\ttranscript\t', gtf_lines)]
tx2gene <- data.frame( TXNAME = sub('.*transcript_id "([^"]+)".*', '\\1', tx_lines), GENEID = sub('.*gene_id "([^"]+)".*', '\\1', tx_lines)) |> dplyr::distinct() |> dplyr::mutate( # Strip version numbers to match --gencode flag in salmon index TXNAME = sub("\\.\\d+$", "", TXNAME), GENEID = sub("\\.\\d+$", "", GENEID) )
dim(tx2gene) # should be ~250,000 rows for GENCODE 45 humanhead(tx2gene)Step 5: Import with tximport and Feed into DESeq2
tximport by Soneson, Love, and Robinson (2015, F1000Research) handles the transcript-to-gene aggregation and produces a length-correction offset matrix that accounts for differential isoform usage across samples. This offset is important: without it, a gene that switches from a long isoform to a short isoform may appear downregulated in a naive count analysis simply because shorter transcripts generate fewer fragments at equal expression.
library(tximport)library(DESeq2)
# List the quant.sf files in sample order matching your metadataquant_files <- file.path( "salmon_output", c("sample_01", "sample_02", "sample_03", "sample_04", "sample_05", "sample_06"), "quant.sf")names(quant_files) <- c("sample_01", "sample_02", "sample_03", "sample_04", "sample_05", "sample_06")
# Verify all files exist before importingstopifnot(all(file.exists(quant_files)))
# Import with tximport# type = "salmon" reads the quant.sf format# tx2gene: transcript-to-gene mapping built above# countsFromAbundance = "lengthScaledTPM": recommended for DESeq2 (offsets for isoform switching)txi <- tximport( quant_files, type = "salmon", tx2gene = tx2gene, countsFromAbundance = "lengthScaledTPM", ignoreTxVersion = TRUE # matches --gencode stripping in index)
# Inspect the outputdim(txi$counts) # genes x samplesdim(txi$abundance) # same: TPM valuessummary(colSums(txi$counts)) # library sizes should be in the millions
# Load metadatametadata <- data.frame( condition = factor(c("control", "control", "control", "treated", "treated", "treated"), levels = c("control", "treated")), row.names = names(quant_files))
# Build DESeqDataSet directly from tximport output# Note: use DESeqDataSetFromTximport, NOT DESeqDataSetFromMatrixdds <- DESeqDataSetFromTximport( txi = txi, colData = metadata, design = ~ condition)
# Pre-filterdds <- dds[rowSums(counts(dds)) >= 10, ]
# Run DESeq2dds <- DESeq(dds)res <- results(dds, name = "condition_treated_vs_control")summary(res)The DESeqDataSetFromTximport function (not DESeqDataSetFromMatrix) is the correct constructor when coming from tximport. It automatically incorporates the offset matrix that corrects for effective gene length differences across samples. Using DESeqDataSetFromMatrix on the txi$counts object directly discards this offset, removing one of the key advantages of the Salmon-tximport pipeline.
The post DESeq2 vignette on tximport integration at Bioconductor, maintained by Michael Love’s group, is the authoritative reference for this specific workflow. The tximport vignette by Soneson and Love documents the countsFromAbundance options and their statistical implications. Both are worth bookmarking.
Final Sanity Checks
Before proceeding to differential expression, run three sanity checks:
# 1. Are library sizes consistent across samples?# Large discrepancies (>5x) warrant investigationcolSums(counts(dds)) |> sort()
# 2. Do samples cluster by condition on PCA?vsd <- vst(dds, blind = TRUE)plotPCA(vsd, intgroup = "condition")
# 3. Is the mapping rate consistent with expected library quality?# (already checked in bash step above, but confirm here)# If any sample has < 80% mapping rate, investigate before publishingNotchBio uses this exact Salmon configuration by default: decoy-aware indexing with --gencode flag, quantification with --validateMappings, --gcBias, and --seqBias, tximport with lengthScaledTPM, and DESeqDataSetFromTximport as the DESeq2 entry point. The mapping rate for every sample is reported in the run summary before differential expression begins, and any sample below 75 percent mapping rate triggers a warning that blocks the analysis until you confirm how to proceed.
Related Reading
Further reading
Read another related post
Build a Counts Matrix from featureCounts and Salmon
Python tutorial: parse featureCounts output, aggregate Salmon quant.sf, build a tx2gene map, and save a DESeq2-ready integer count matrix with pandas.
TutorialHow to Run STAR Alignment for Bulk RNA-Seq (Step-by-Step)
Complete STAR tutorial: download genome and GTF, build an index with the right sjdbOverhang, run paired-end alignment, and load GeneCounts into R for DESeq2.
TutorialHow to Build a Salmon Index and Quantify Bulk RNA-Seq Reads
Step-by-step Salmon tutorial: download GENCODE references, build a decoy-aware index, run salmon quant with gcBias and seqBias, and verify mapping rates.