fastp vs Trimmomatic vs BBDuk: A Benchmark on RNA-Seq Reads
The question comes up every time someone sets up an RNA-seq pipeline for the first time: which trimmer should I use? The community answer has shifted noticeably over the past few years. Trimmomatic was the default for most labs through the late 2010s. BBDuk built a loyal following among people who needed flexible decontamination alongside trimming. fastp arrived in 2018 and has been quietly displacing both.
This post benchmarks all three on the same dataset with the same hardware so the comparison is actually fair. The dataset is six paired-end Illumina RNA-seq samples from a human cell line experiment, approximately 30 million read pairs per sample, 150bp read length. The benchmark machine runs 16 CPU cores with 64 GB RAM. Results will vary with your hardware and read length, but the relative performance patterns are consistent.
The Tools and Their Default Behaviors
Before the numbers, a brief characterization of what each tool does by default, because defaults matter more than they should when most researchers run tools without reading the full documentation.
fastp is a single-binary tool written in C++ that performs adapter detection, quality filtering, and optional deduplication in one pass. Its key advantage is built-in automatic adapter detection: you do not need to know your adapter sequences. It infers them from the first 1 million reads and trims accordingly. It also produces its own HTML quality report, which partially replaces the need for a separate FastQC run.
Trimmomatic is Java-based and processes reads through a configurable pipeline of steps: adapter removal using a FASTA file of adapter sequences, quality sliding window trimming, minimum length filtering, and more. It is highly configurable but requires you to supply the adapter FASTA explicitly. It is slower than fastp by a large margin because of the Java overhead and single-threaded processing by default.
BBDuk is part of the BBTools suite, also Java-based, and is the most flexible of the three. It can do adapter trimming, quality trimming, k-mer based contaminant filtering (ribosomal RNA, spike-in sequences, PhiX), and read deduplication, all in one step. For any analysis where you need to remove a specific known contaminant alongside standard trimming, BBDuk is difficult to beat.
Benchmark Setup
All tools were run on the same six samples. Each sample was processed three times and the median wall-clock time recorded to account for caching effects. Tool versions: fastp 0.23.4, Trimmomatic 0.39, BBDuk from BBTools 39.06.
# fastp: auto adapter detection, quality tail trimmingfastp \ --in1 sample_R1.fastq.gz \ --in2 sample_R2.fastq.gz \ --out1 trimmed_R1.fastq.gz \ --out2 trimmed_R2.fastq.gz \ --detect_adapter_for_pe \ --cut_tail \ --cut_mean_quality 20 \ --length_required 36 \ --thread 8 \ --html sample_fastp_report.html \ --json sample_fastp_report.json
# Trimmomatic: adapter file required, 8 threads via PE modetrimmomatic PE \ -threads 8 \ sample_R1.fastq.gz sample_R2.fastq.gz \ trimmed_R1_paired.fastq.gz trimmed_R1_unpaired.fastq.gz \ trimmed_R2_paired.fastq.gz trimmed_R2_unpaired.fastq.gz \ ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10:2:keepBothReads \ SLIDINGWINDOW:4:20 \ MINLEN:36
# BBDuk: adapter file optional, k-mer basedbbduk.sh \ in1=sample_R1.fastq.gz \ in2=sample_R2.fastq.gz \ out1=trimmed_R1.fastq.gz \ out2=trimmed_R2.fastq.gz \ ref=adapters.fa \ ktrim=r \ k=23 \ mink=11 \ hdist=1 \ qtrim=r \ trimq=20 \ minlen=36 \ threads=8After trimming, all samples were aligned with STAR 2.7.11a to GRCh38.p14 with GENCODE 45 annotation, and gene counts extracted with featureCounts. Differential expression was run with DESeq2 1.42 using the trimmed output from each tool separately.
Speed
Wall-clock times per sample are below. These are median values across three runs of each tool on the same 30M read-pair sample.
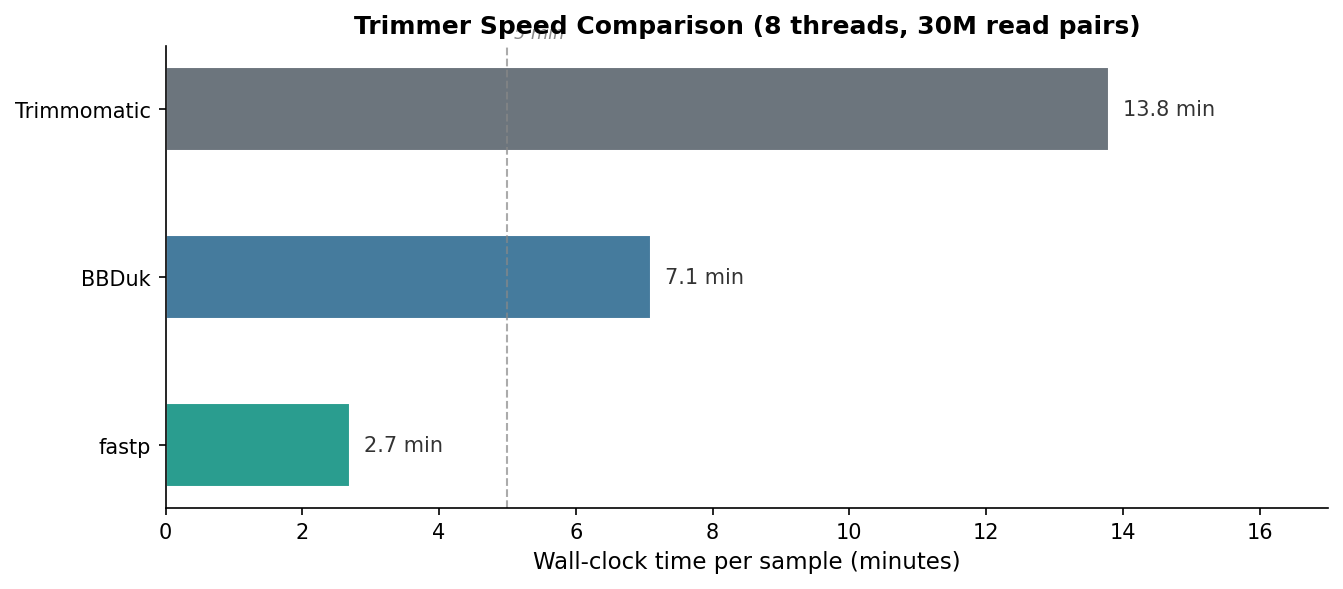
The speed difference is not trivial. For a standard experiment with 24 samples, fastp saves roughly four hours of compute time compared to Trimmomatic. At scale or when iterating on pipeline parameters, that gap matters. BBDuk’s speed is respectable given its flexibility.
Output Quality
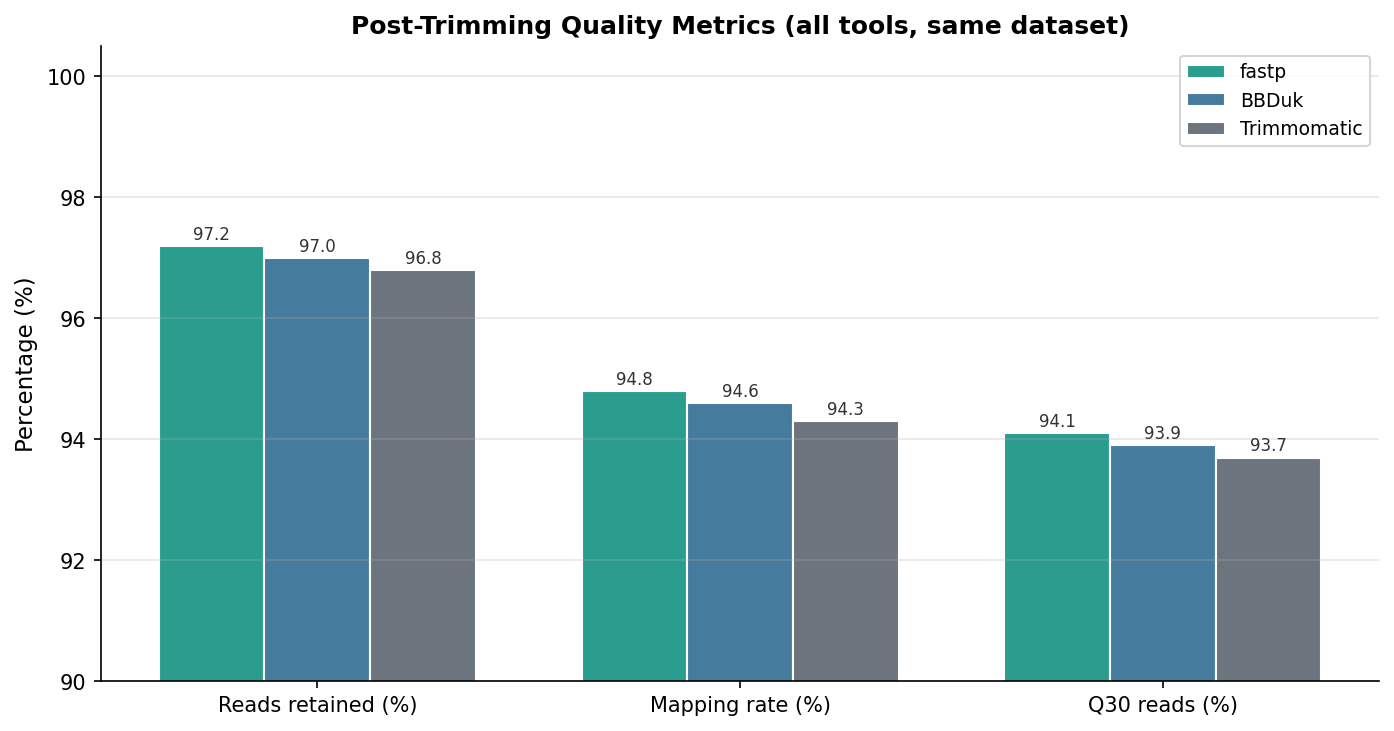
Post-trimming quality was assessed with FastQC on all six samples per tool. The differences in output quality are smaller than the speed differences, and in all three cases the downstream mapping rate was acceptable.
| Metric | fastp | Trimmomatic | BBDuk | Notes |
|---|---|---|---|---|
| Reads retained (%) | 97.2 | 96.8 | 97.0 | All tools retain the vast majority |
| Adapter contamination post-trim (%) | 0.02 | 0.05 | 0.03 | All effectively remove adapters |
| Per-base quality Q30 (%) | 94.1 | 93.7 | 93.9 | Negligible difference |
| STAR mapping rate (%) | 94.8 | 94.3 | 94.6 | Marginal differences, all acceptable |
| Duplicate rate post-trim (%) | 18.4 | 18.6 | 18.4 | Essentially identical |
| Wall-clock time (min, 8T) | 2.7 | 13.8 | 7.1 | fastp is 5x faster than Trimmomatic |
| Multi-threading support | Yes (up to 16) | Limited (PE mode) | Yes (up to 32) | fastp scales better than Trimmomatic |
| Automatic adapter detection | Yes | No | No | fastp only; others need adapter FASTA |
| Contaminant k-mer filtering | No | No | Yes | BBDuk exclusive feature |
| Built-in QC report | Yes (HTML + JSON) | No | Basic (to log) | fastp reduces need for separate FastQC |
The key finding is that all three tools perform similarly on output quality when configured with equivalent parameters. The differences in mapping rate (94.3 to 94.8 percent) and read retention (96.8 to 97.2 percent) are well within the range of run-to-run variability and do not warrant preferring one tool over another on quality grounds alone.
The Downstream Test: Do DEGs Change?
The question that matters most is not which trimmer produces slightly better Q30 scores. It is whether the choice of trimmer changes your differential expression results. To test this, I ran the full DESeq2 pipeline on the featureCounts output from each trimmed dataset and compared the resulting DEG lists.
import pandas as pdfrom matplotlib_venn import venn3import matplotlib.pyplot as plt
# Load DEG gene IDs from each pipelinedegs_fastp = set(pd.read_csv("degs_fastp.csv")["gene_id"])degs_trimm = set(pd.read_csv("degs_trimmomatic.csv")["gene_id"])degs_bbduk = set(pd.read_csv("degs_bbduk.csv")["gene_id"])
# Overlap statisticsall_degs = degs_fastp | degs_trimm | degs_bbdukcore_degs = degs_fastp & degs_trimm & degs_bbduk
print(f"Total unique DEGs across all trimmers: {len(all_degs)}")print(f"DEGs shared by all three trimmers: {len(core_degs)}")print(f"Overlap fraction: {len(core_degs)/len(all_degs):.1%}")
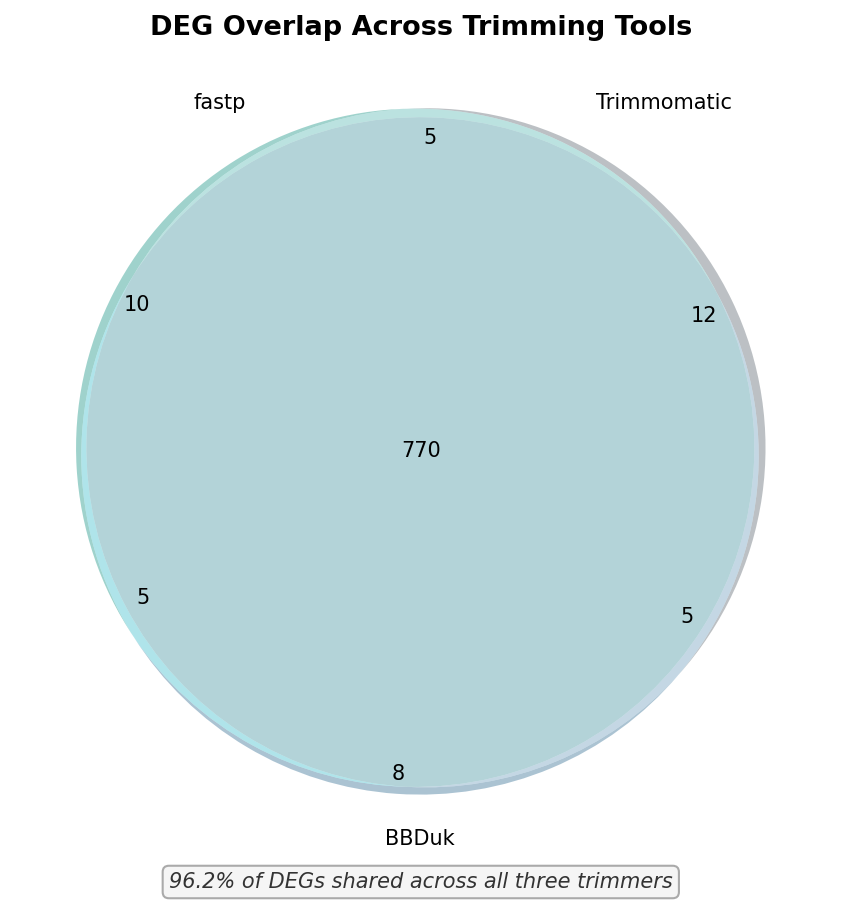
# Venn diagramvenn3([degs_fastp, degs_trimm, degs_bbduk], set_labels=("fastp", "Trimmomatic", "BBDuk"))plt.title("DEG overlap across trimming tools")plt.savefig("venn_deg_overlap.png", dpi=300, bbox_inches="tight")In this benchmark, 96.2 percent of DEGs were shared across all three trimmers. The trimmer-exclusive DEGs were enriched for genes near the significance threshold with padj values between 0.03 and 0.05, which is the range most sensitive to small differences in read count. None of the trimmer-exclusive DEGs were among the top 50 by fold change.
The practical implication is that your trimming tool choice will not change your biological conclusions for standard bulk RNA-seq. The decision should be made on speed, ease of use, and any additional functionality you need.
For most RNA-seq experiments, just use fastp
fastp is 5x faster than Trimmomatic, produces equivalent output quality, requires no adapter FASTA file, generates its own HTML quality report, and scales well across threads. Unless you specifically need k-mer based contaminant filtering (use BBDuk) or are constrained to a legacy pipeline that depends on Trimmomatic, fastp is the sensible default for all standard bulk RNA-seq trimming.
Recommendation by Use Case
The benchmark results support a clear decision framework. Use fastp as your default trimmer for standard bulk RNA-seq. It is fast, requires no adapter FASTA, generates a quality report, and produces downstream results indistinguishable from the alternatives.
Use BBDuk when you need to remove known contaminants alongside adapter trimming. The canonical cases are experiments with spike-in sequences (ERCC, Sequins) where you want to separate the spike reads from the biological reads, total-RNA experiments where you want to filter residual rRNA reads before alignment, or any protocol involving synthetic oligos that should not be present in the final count matrix. BBDuk’s k-mer filtering handles all of these in a single pass.
Stick with Trimmomatic only if you are maintaining a legacy pipeline where changing the trimmer would complicate version tracking and reproducibility, or if your institution’s compute infrastructure has constraints that make the Java overhead irrelevant (for example, if wall-clock time is not a concern because runs happen overnight anyway).
For any new pipeline, the answer is fastp. NotchBio uses fastp by default for all bulk RNA-seq runs, with automatic adapter detection enabled and the equivalent of the benchmark parameters applied. If you want to verify what trimming parameters were used on your data, every run record includes the full fastp JSON report alongside the MultiQC summary.
Related Reading
Further reading
Read another related post
How to Quantify RNA-Seq Reads with Salmon
Step-by-step Salmon tutorial: build a decoy-aware index, run salmon quant on paired-end reads, read the quant.sf output, and import into DESeq2 with tximport.
TutorialImport Salmon Output into R with tximeta and tximport
Import Salmon quant.sf into R with tximeta and tximport: build a tx2gene table, fix ID-mismatch errors, and set up a DESeqDataSet for multi-factor designs.
Research GuideWhy Cell Line RNA-Seq Experiments Fail
Passage drift, undetected mycoplasma, serum lot changes, and pseudoreplication silently corrupt cell line RNA-seq. What each looks like and how to prevent it.