How To Submit RNA-Seq Results That Reviewers Cannot Reject
Reviewer two sent the manuscript back with seven comments. Four of them were about the RNA-seq methods section. Specifically: no version numbers for any of the tools, no statement of which genome assembly was used, the enrichment analysis does not mention whether FDR correction was applied, and the code availability statement says “available from the corresponding author upon reasonable request,” which the reviewer notes is not the same as available.
None of those comments are unfair. All of them reflect minimum reproducibility standards that the field has published guidance on for years. And all of them are entirely preventable, because the information the reviewer is asking for already existed before the paper was submitted. The only question was whether someone decided to include it.
This post is the checklist that prevents reviewer two from writing those four comments. It covers the five most common computational reasons RNA-seq papers come back from review, the pre-submission checklist that catches each one, what belongs in main text versus supplementary, and how to write data and code availability statements that satisfy reviewers at journals from PLOS to Nature.
The Five Most Common Reviewer Rejection Reasons
These five categories account for the large majority of revision requests that relate specifically to the computational analysis in RNA-seq papers. They come from a combination of the published reproducibility literature and consistent patterns in the community.
Missing or unspecified tool versions. A 2021 systematic review published in Briefings in Bioinformatics analyzed 1,000 RNA-seq papers and found that tool version reporting was one of the most consistently absent elements across the field. DESeq2 without a version number is not reproducible. The statistical behavior of the tool changes between major releases. A reviewer who knows the field will ask. A reviewer who does not will not, but the paper will still be irreproducible.
Unspecified reference genome or annotation. “Reads were aligned to the human genome” identifies the species. It does not identify the assembly (GRCh38 versus T2T-CHM13), the patch level (GRCh38.p14), or the annotation release (GENCODE 45 versus Ensembl 111). These choices affect which genes appear in the count matrix, how many reads align, and what the results look like. Reviewers in genomics journals increasingly flag this explicitly.
Missing or incorrectly applied multiple testing correction. The same 2021 study found that a significant fraction of papers either omit the correction method entirely or apply it incorrectly. Writing “genes with p < 0.05 were considered differentially expressed” without specifying whether that is a raw or adjusted p-value is ambiguous in a way that reviewers will not let pass. If the GO enrichment results come from ORA without specifying the background gene set and the correction method, that is a separate but related problem.
Code and data availability statements that do not actually provide access. “Available upon reasonable request” is a statement that the data exist. It is not a data availability statement. Nucleic Acids Research and an increasing number of journals explicitly state that this phrasing is insufficient and can delay review or cause rejection. FASTQ files belong in GEO or SRA. Code belongs in a public repository with a DOI. The DOI is what distinguishes “available” from “maybe available for a while.”
Absent or incomplete pre-filtering criteria. If you removed genes with fewer than 10 counts before running DESeq2, that filtering step is part of the analysis and belongs in the methods section. If you excluded samples based on PCA inspection, the criteria for exclusion need to be stated. Undocumented filtering is the most common source of discrepancy when someone attempts to reproduce a published analysis.
| Rejection reason | What reviewers find | What you need to provide |
|---|---|---|
| Tool versions absent | ”DESeq2 was used” with no version | DESeq2 v1.44.0, R v4.4.1, apeglm v1.24.0 |
| Reference genome unspecified | ”aligned to human genome” | GRCh38.p14, GENCODE release 45 |
| Multiple testing correction unclear | ”p < 0.05” with no clarification | ”Benjamini-Hochberg adjusted p-value < 0.05” |
| GO enrichment background absent | ”enrichment analysis using clusterProfiler” | Background = all genes with non-NA padj from DESeq2 |
| Code unavailable | ”available from corresponding author” | GitHub repository with a Zenodo DOI |
| FASTQ data unavailable | ”data available on request” | GEO accession number (GSExxxxxxx) |
| Filtering criteria absent | no mention of pre-filtering | ”Genes with fewer than 10 counts were excluded” |
| Enrichment method unspecified | ”pathway analysis was performed" | "ORA using enrichGO with BH correction; GSEA using gseGO with Wald statistic ranking” |
The Pre-Submission Checklist
Running through this list before hitting submit takes less than twenty minutes and prevents the majority of computational revision requests. Every item on it corresponds directly to one of the rejection reasons above.
The pre-submission checklist: run this before every submission
Tool versions: every tool named in the methods section has an exact version number. Reference genome: the assembly name, patch level, and annotation release are all specified with a stable identifier or URL. Statistical thresholds: every p-value threshold states whether it applies to raw or adjusted values, and the correction method is named explicitly. Enrichment analysis: the method (ORA or GSEA), the background gene set (how defined), the significance threshold, and the correction method are all present. Pre-filtering: any gene or sample exclusion is described with the explicit criterion that was applied. Code: deposited in a public repository (GitHub, GitLab, or Bitbucket) with a DOI obtained from Zenodo, Figshare, or the Open Science Framework. FASTQ data: submitted to GEO or SRA before the paper is submitted, with an accession number included in the submission.
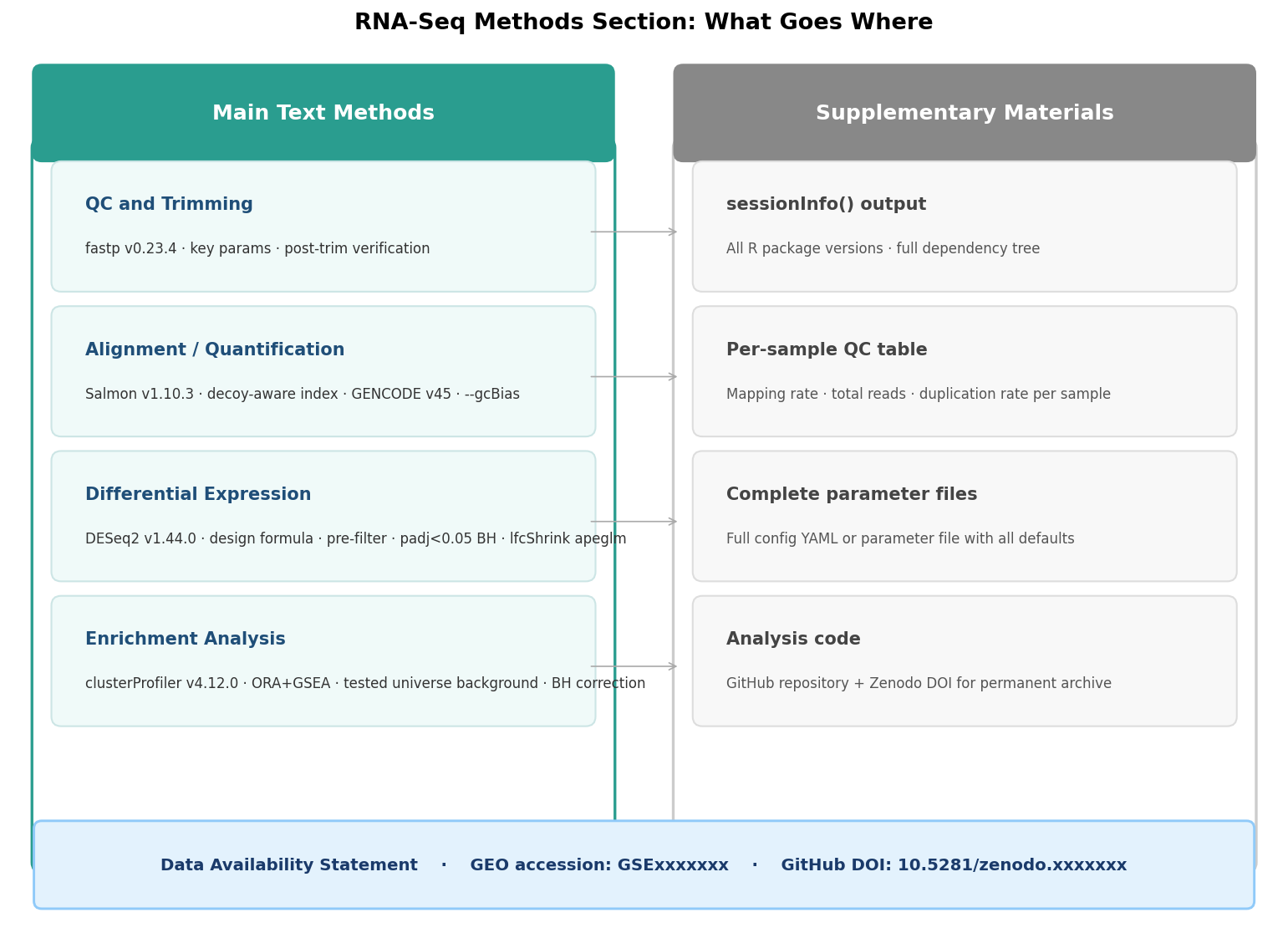
What Goes Where: Main Text Versus Supplementary
The structure of an RNA-seq methods section that satisfies reviewers follows a consistent pattern. Main text covers everything a reader needs to evaluate and potentially reproduce the primary analysis at a high level. Supplementary covers the detail that would interrupt the narrative but that a careful reader needs to fully reproduce the work.
In the main text methods:
One paragraph per analysis stage, in execution order. Quality control and trimming: tool, version, key non-default parameters. Alignment or quantification: tool, version, reference genome, annotation release. Differential expression: design formula written out, pre-filtering criterion, significance thresholds with correction method named. Enrichment analysis: ORA or GSEA, background definition, correction method, significance threshold. Any custom steps or deviations from standard practice.
In supplementary methods:
Full sessionInfo() output from R, or the complete conda environment file. Complete parameter settings for any tool where you changed defaults beyond what fits in the main text. The exact commands or script used for each pipeline step, if space allows. Sample-level QC metrics as a supplementary table (mapping rate, total reads mapped, duplication rate per sample). Any exclusion decisions with justification.
Not in the paper at all:
Default parameter values for tools you ran with no changes, when the default behavior is documented in the tool’s primary citation. Algorithmic descriptions of tools you used but did not modify. Version history of the analysis as you developed it during the project.
Code and Data Availability Statements That Work
The two most common problems with availability statements are vagueness (“available upon request”) and missing identifiers (no accession number, no DOI). Both make reviewers ask follow-up questions during revision, which delays publication.
A data availability statement that works:
“RNA-seq data (FASTQ files and processed count matrices) have been deposited in the Gene Expression Omnibus under accession number GSExxxxxxx and are publicly accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSExxxxxxx. Processed data are available immediately; raw FASTQ files will be released upon publication.”
A code availability statement that works:
“All analysis code is available at https://github.com/lab/project-name (archived version: DOI 10.5281/zenodo.xxxxxxx). The repository contains all scripts used for quality control, quantification, differential expression analysis, and enrichment analysis, along with a README describing the execution order and dependencies.”
The GEO submission should be completed before the paper is submitted, not after acceptance. Most journals now require the accession number to be included in the submitted manuscript. If the data are embargoed until publication, GEO provides a private reviewer access link that can be included in the cover letter.
The Zenodo DOI is obtained by creating a GitHub release and connecting the repository to Zenodo through their GitHub integration. This takes about fifteen minutes and turns a GitHub repository link, which can disappear if the repository is renamed or deleted, into a permanent citable archive.
The Case for Starting With a Reproducible Platform
Every item on the checklist above represents information that already existed at the time the analysis was run. The challenge is capturing it at that moment rather than trying to reconstruct it from memory six months later when the manuscript is being prepared.
A platform that records this information automatically as part of every run changes the preparation of the methods section from a reconstruction task to a retrieval task. The version numbers are in the run record. The parameter settings are in the run record. The sample-to-condition mapping is in the run record. The methods paragraph is generated from the run record.
NotchBio generates a complete methods paragraph for every RNA-seq run, including tool versions, parameter choices, reference genome version, and formatted citations for each tool in the pipeline. Every run produces a permalink that captures all five reproducibility layers described earlier in this series. If a reviewer asks what version of DESeq2 was used, the answer is the permalink. If a reviewer asks whether lfcShrink was applied, the answer is in the methods text that the platform generated when the run completed.
The pre-submission checklist above should take twenty minutes to run before you submit. If your analysis was run through NotchBio, most of that checklist is already satisfied before you start writing the methods section. That is not a minor convenience. When you are writing a paper, the difference between “I need to reconstruct what I did eight months ago” and “the methods text was generated when I ran the analysis” is the difference between a straightforward submission and a stressful one.
If you are planning a new RNA-seq experiment and want the methods section to write itself, start your next analysis at notchbio.app. The reviewer comments about version numbers and missing correction methods are preventable. This is how you prevent them.
Related Reading
Further reading
Read another related post
PyDESeq2 Tutorial: Differential Expression in Python
The complete PyDESeq2 reference in Python: DeseqDataSet, DeseqStats, apeglm shrinkage, multi-factor designs, multiple contrasts, and pandas result filtering.
TutorialHow to Run DESeq2: From Count Matrix to Results
Step-by-step DESeq2 in R: build a DESeqDataSet, understand size factors and dispersion, run DESeq(), interpret the results columns, then shrink and filter DEGs.
TutorialSet Up a Bulk RNA-Seq Environment on Ubuntu and macOS
Install Miniforge, conda, bioconda, R 4.4, and DESeq2 for bulk RNA-seq: reproducible environments, version pinning, and fixes for common install errors.