Differential Expression Analysis in Python with PyDESeq2: A Complete Tutorial
R has owned bulk RNA-seq differential expression for a decade. DESeq2 is the dominant tool, it lives in R, and most tutorials assume you are comfortable with R data structures. If your analysis stack is Python, the standard advice has been to either write a subprocess call to R or re-implement the normalization yourself and accept inferior statistics.
PyDESeq2 changes this. Published in Bioinformatics in 2023, it is a from-scratch Python reimplementation of the DESeq2 workflow, written in pure Python using NumPy and SciPy, with no R dependency anywhere in the call stack (Muzellec et al., 2023). It is part of the scverse ecosystem alongside Scanpy and AnnData, which means it integrates naturally with the Python single-cell and multi-omics tooling that has become standard in modern labs.
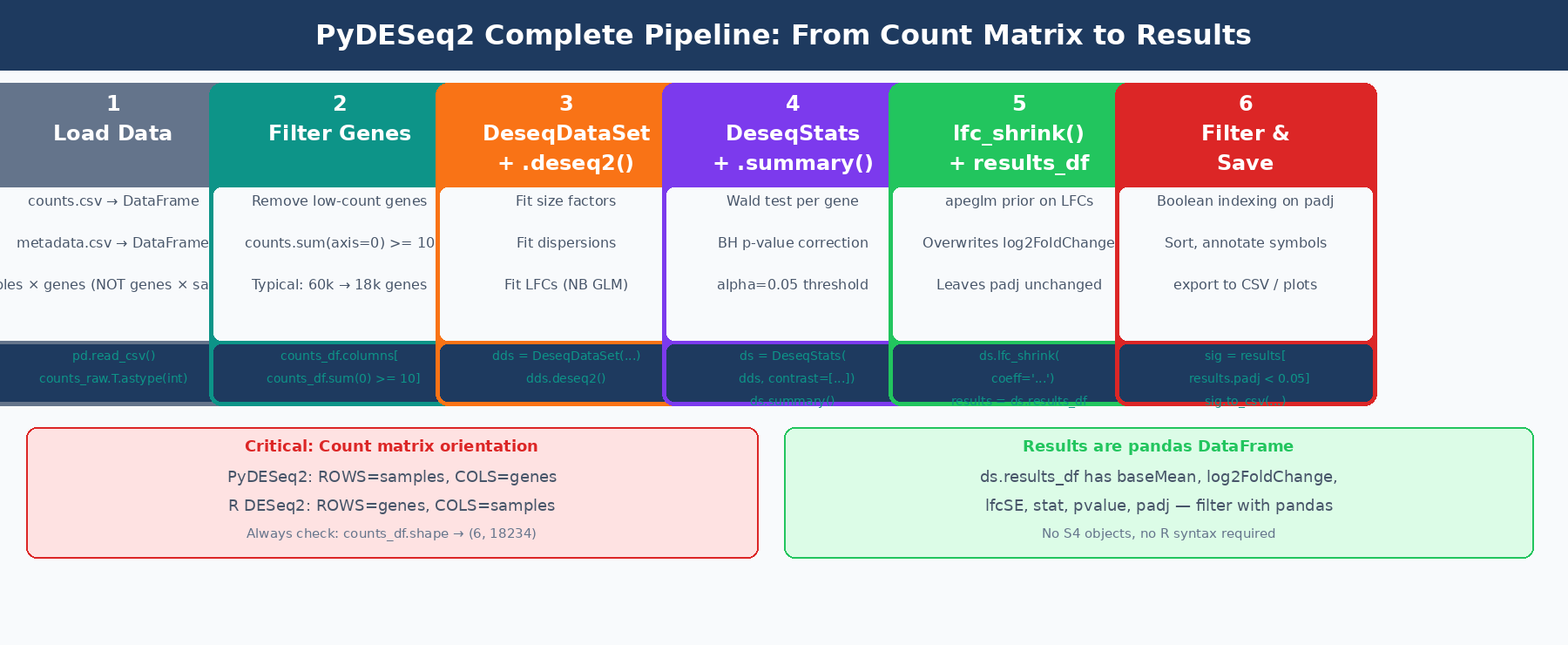
This tutorial walks through the complete PyDESeq2 workflow from scratch: installation, input preparation, model fitting, statistical testing, LFC shrinkage, and saving results. It also covers the critical ways PyDESeq2 differs from DESeq2 in R, which are the differences that will catch you if you switch without reading the docs.
For the R-native mental model behind the same statistics, pair this with How DESeq2 Actually Works (Without the Math Overload) and How to Run DESeq2: A Complete Walkthrough from Count Matrix to Results. If your starting point is still the input table, What Is a Count Matrix and Why Does It Matter is the prerequisite refresher.
What Is PyDESeq2 and How Does It Compare to DESeq2 in R?
PyDESeq2 implements the same negative binomial GLM model as DESeq2. The workflow is identical: estimate size factors, estimate and shrink gene-wise dispersions, fit the GLM, and perform Wald tests. The published benchmarks on eight TCGA datasets show that PyDESeq2 returns very similar sets of significant genes and pathways to DESeq2, while achieving higher model likelihood for dispersion and LFC parameters on the vast majority of genes, at comparable speeds for large cohorts (PMC10502239).
The practical differences that matter for day-to-day use:
Count matrix orientation is transposed. In R DESeq2, the count matrix is genes × samples (rows are genes). In PyDESeq2, the count matrix must be samples × genes (rows are samples, columns are genes). This is the most common source of silent failure when porting from R.
Two-class structure instead of one. DESeq2 in R wraps everything in a single DESeqDataSet. PyDESeq2 separates modeling from testing: DeseqDataSet fits dispersions and LFCs, DeseqStats runs the Wald test and shrinkage.
Results are a pandas DataFrame. Instead of an S4 object you query with accessor functions, ds.results_df is a standard pandas DataFrame you can filter, sort, merge, and plot with the full Python data science toolchain.
Currently implements Wald tests only. Likelihood Ratio Tests (LRT), which are useful for time-course and multi-level experiments, are not yet implemented as of version 0.5. Check the GitHub releases page for current feature status.
| Feature | DESeq2 (R) | PyDESeq2 (Python) |
|---|---|---|
| Language | R | Python (NumPy, SciPy) |
| Count matrix orientation | Genes × samples | Samples × genes |
| Main objects | DESeqDataSet | DeseqDataSet + DeseqStats |
| Results format | S4 DESeqResults object | pandas.DataFrame (ds.results_df) |
| Wald tests | Yes | Yes |
| Likelihood Ratio Tests | Yes | Not yet |
| apeglm LFC shrinkage | Yes | Yes |
| Integration with Python ecosystem | Via rpy2 only | Native |
| Scverse/AnnData compatible | No | Yes |
How to Install PyDESeq2
# Recommended: in a clean conda environmentconda create -n pydeseq2 python=3.11 -yconda activate pydeseq2
# Install from PyPIpip install pydeseq2
# Install additional tools used in this tutorialpip install pandas numpy matplotlib seaborn scikit-learn scipyVerify the installation:
import pydeseq2print(pydeseq2.__version__)# 0.5.4 (or later)PyDESeq2 is also available through Bioconda (conda install -c bioconda pydeseq2) and is included in the scverse conda channel. Check the PyDESeq2 GitHub for the latest installation instructions, as the package has moved between organizations since its initial release.
How to Prepare Your Count Matrix for PyDESeq2
This is the step most tutorials gloss over, and it is where most errors originate. PyDESeq2 requires:
A count matrix with samples as rows and genes as columns. This is the opposite of the R convention.
Values must be non-negative integers (raw counts, not TPM or normalized values).
The index of the count DataFrame must match the index of the metadata DataFrame exactly.
import pandas as pdimport numpy as np
# ── Option A: Load from featureCounts CSV ──────────────────────# featureCounts typically outputs genes as rows, samples as columnscounts_raw = pd.read_csv( "results/counts/feature_counts.csv", index_col = 0 # gene IDs are in column 0)
# featureCounts format: genes x samples → transpose for PyDESeq2counts_df = counts_raw.T
print(f"Count matrix shape: {counts_df.shape}")# (6, 18234) ← rows=samples, columns=genes
# Verify integer countsassert (counts_df >= 0).all().all(), "Counts contain negative values"counts_df = counts_df.astype(int)
# ── Option B: Load from Salmon quant.sf files (Python alternative to tximport) ──# For each sample, load NumReads column and build the matriximport osfrom pathlib import Path
sample_dirs = sorted(Path("results/salmon").glob("*_quant"))sample_names = [d.name.replace("_quant", "") for d in sample_dirs]
counts_dict = {}for name, sdir in zip(sample_names, sample_dirs): quant = pd.read_csv( sdir / "quant.sf", sep = "\t", index_col = "Name", usecols = ["Name", "NumReads"] ) # Round fractional estimated counts to integers counts_dict[name] = quant["NumReads"].round().astype(int)
counts_df = pd.DataFrame(counts_dict).T # samples x genesprint(f"Loaded {counts_df.shape[0]} samples, {counts_df.shape[1]} transcripts")
# ── Build metadata DataFrame ────────────────────────────────────metadata = pd.DataFrame({ "condition": ["control", "control", "control", "treated", "treated", "treated"], "batch": ["A", "A", "B", "A", "A", "B"]}, index = sample_names)
# Condition must be a string or categorical, not numericmetadata["condition"] = metadata["condition"].astype(str)metadata["batch"] = metadata["batch"].astype(str)
# Verify alignment: sample order must match in both DataFramesassert list(counts_df.index) == list(metadata.index), \ "Sample order mismatch between counts and metadata"
print(metadata)# condition batch# ctrl_1 control A# ctrl_2 control A# ctrl_3 control B# treat_1 treated A# treat_2 treated A# treat_3 treated BHow to Filter Low-Count Genes Before Running PyDESeq2
Low-count genes add noise without contributing statistical power. Filter them out before model fitting:
# Keep genes with at least 10 total counts across all samplesgenes_to_keep = counts_df.columns[counts_df.sum(axis=0) >= 10]counts_df = counts_df[genes_to_keep]
print(f"Genes after filtering: {counts_df.shape[1]}")# Before: ~60,000 genes After: ~18,000-22,000 genes (human, GENCODE)This threshold is a starting point. For experiments with more replicates, you can be more stringent (e.g., requiring at least 10 counts in at least half the samples). For small experiments, be conservative.
The count matrix orientation in PyDESeq2 is samples × genes, not genes × samples
If you pass a genes × samples matrix to DeseqDataSet, the model will treat each gene as a sample and each sample as a gene. You will not get an error, but your results will be completely meaningless. Always check counts_df.shape before proceeding: the number of rows should be your sample count (typically 6 to 60) and the number of columns should be your gene count (typically 10,000 to 25,000).
How to Run Differential Expression Analysis with PyDESeq2
The PyDESeq2 workflow separates into two steps: DeseqDataSet for model fitting, DeseqStats for hypothesis testing.
Step 1: Fit the Model with DeseqDataSet
from pydeseq2.dds import DeseqDataSetfrom pydeseq2.default_inference import DefaultInference
# DefaultInference controls parallelizationinference = DefaultInference(n_cpus=8)
# Create and fit the DeseqDataSetdds = DeseqDataSet( counts = counts_df, metadata = metadata, design = "~condition", # statistical design formula refit_cooks = True, # refit genes with Cook's distance outliers inference = inference)
# Fit the model: estimates size factors, dispersions, and LFCsdds.deseq2()The dds.deseq2() call runs three steps and prints progress for each:
Fitting size factors......done in 0.02 seconds.Fitting dispersions......done in 4.31 seconds.Fitting LFCs......done in 3.87 seconds.Replacing 2 outliers.Refitting 2 outliers....done in 0.14 seconds.Size factors are accessible at dds.obsm["size_factors"]. Dispersion estimates are in dds.varm["dispersions"]. You can plot the dispersion curve:
dds.plot_dispersions()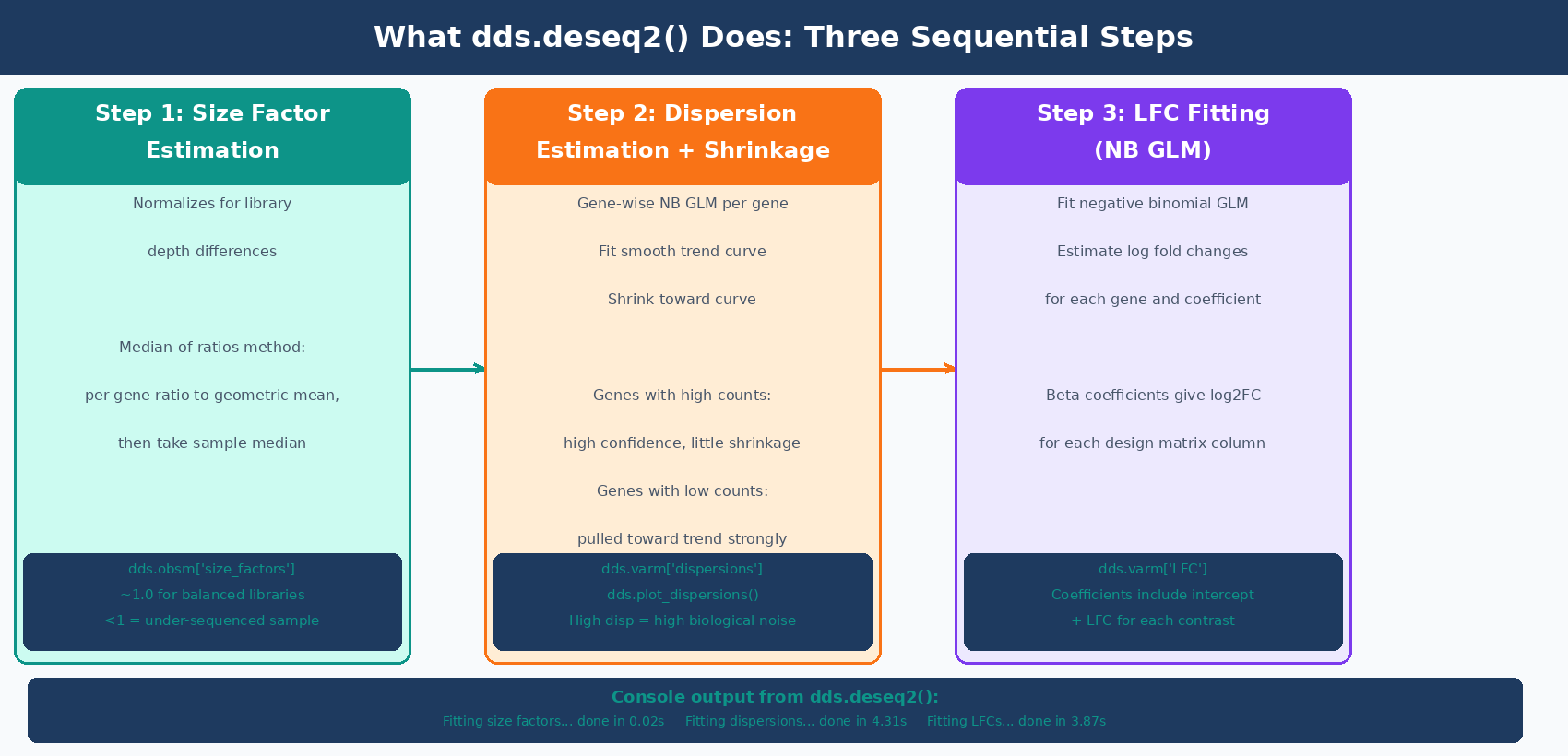
Step 2: Run Statistical Tests with DeseqStats
from pydeseq2.ds import DeseqStats
# Create DeseqStats object with a contrast# contrast = ["variable", "tested_level", "reference_level"]ds = DeseqStats( dds, contrast = ["condition", "treated", "control"], alpha = 0.05, # significance threshold for summary() cooks_filter = True, # filter genes with Cook's outliers independent_filter = True, # independent filtering to improve power inference = inference)
# Run the Wald test and BH multiple testing correctionds.summary()ds.summary() prints:
Running Wald tests......done in 2.14 seconds.Log2 fold change & Wald test p-value: condition treated vs control
baseMean log2FoldChange lfcSE stat pvalue padjBRCA1 1247.302 -1.432 0.124 -11.558 5.42e-31 1.13e-27MYC 3890.441 2.891 0.089 32.483 1.21e-231 2.51e-227...
Number of genes with padj < 0.05: 1842The results are now in ds.results_df, a standard pandas DataFrame.
How to Apply LFC Shrinkage in PyDESeq2
Like DESeq2 in R, the log2FoldChange column in ds.results_df before shrinkage contains MLE estimates that are noisy for lowly expressed genes. Apply lfc_shrink() to produce apeglm-shrunken estimates for visualization and ranking:
# Apply apeglm LFC shrinkage# The coeff argument specifies which model coefficient to shrinkds.lfc_shrink(coeff="condition_treated_vs_control")
# lfc_shrink() overwrites ds.results_df in place# Verify shrinkage was applied:print(ds.shrunk_LFCs) # TrueImportant: In PyDESeq2, lfc_shrink() modifies ds.results_df in place, overwriting the log2FoldChange column. If you need to compare shrunk and unshrunk values, save the unshrunk DataFrame before calling lfc_shrink():
# Save unshrunk results before shrinkageresults_unshrunk = ds.results_df.copy()
# Apply shrinkage (modifies ds.results_df in place)ds.lfc_shrink(coeff="condition_treated_vs_control")
# Now ds.results_df contains shrunk LFCsresults_shrunk = ds.results_df.copy()
# Compare a few genesprint(results_unshrunk[["log2FoldChange"]].head())print(results_shrunk[["log2FoldChange"]].head())lfc_shrink() changes log2FoldChange but not padj
Like in R, apeglm shrinkage in PyDESeq2 modifies only the log2FoldChange and lfcSE columns. The pvalue and padj columns remain unchanged. Use the shrunk DataFrame for volcano plots, MA plots, and GSEA input. Report significance based on padj regardless of whether shrinkage was applied.
How to Filter and Save PyDESeq2 Results
Since ds.results_df is a pandas DataFrame, filtering and saving uses standard pandas operations:
# Access the results DataFrameresults = ds.results_df.copy()
print(f"Total genes in results: {len(results)}")print(f"Genes with NA padj (filtered by DESeq2): {results['padj'].isna().sum()}")
# Filter to significant DEGssig_degs = results[ (results["padj"] < 0.05) & (results["log2FoldChange"].abs() > 1.0) # 2-fold change].copy()
print(f"Significant DEGs (padj<0.05, |LFC|>1): {len(sig_degs)}")print(f" Upregulated: {(sig_degs['log2FoldChange'] > 0).sum()}")print(f" Downregulated: {(sig_degs['log2FoldChange'] < 0).sum()}")
# Sort by significancesig_degs = sig_degs.sort_values("padj")
# Save all genes (for GSEA — needs full ranked list)results.sort_values("log2FoldChange", ascending=False).to_csv( "results/pydeseq2_all_genes_ranked.csv")
# Save significant DEGssig_degs.to_csv("results/pydeseq2_significant_degs.csv")
print("\nTop 10 significant DEGs:")print(sig_degs.head(10)[["baseMean", "log2FoldChange", "padj"]])Adding Gene Symbols to the Results Table
PyDESeq2 results use the same gene IDs as the count matrix columns. If you used Ensembl IDs, map them to symbols using mygene:
import mygene
# Install with: pip install mygenemg = mygene.MyGeneInfo()
# Strip version numbers from ENSEMBL IDsgene_ids_clean = [g.split(".")[0] for g in results.index]
# Query gene symbols in bulk (much faster than row-by-row)gene_info = mg.querymany( gene_ids_clean, scopes = "ensembl.gene", fields = "symbol", species = "human", as_dataframe = True)
# Map symbols back to resultssymbol_map = gene_info["symbol"].drop_duplicates()results["gene_id"] = gene_ids_cleanresults["symbol"] = results["gene_id"].map(symbol_map)
print(f"Genes with symbol mapping: {results['symbol'].notna().sum()} / {len(results)}")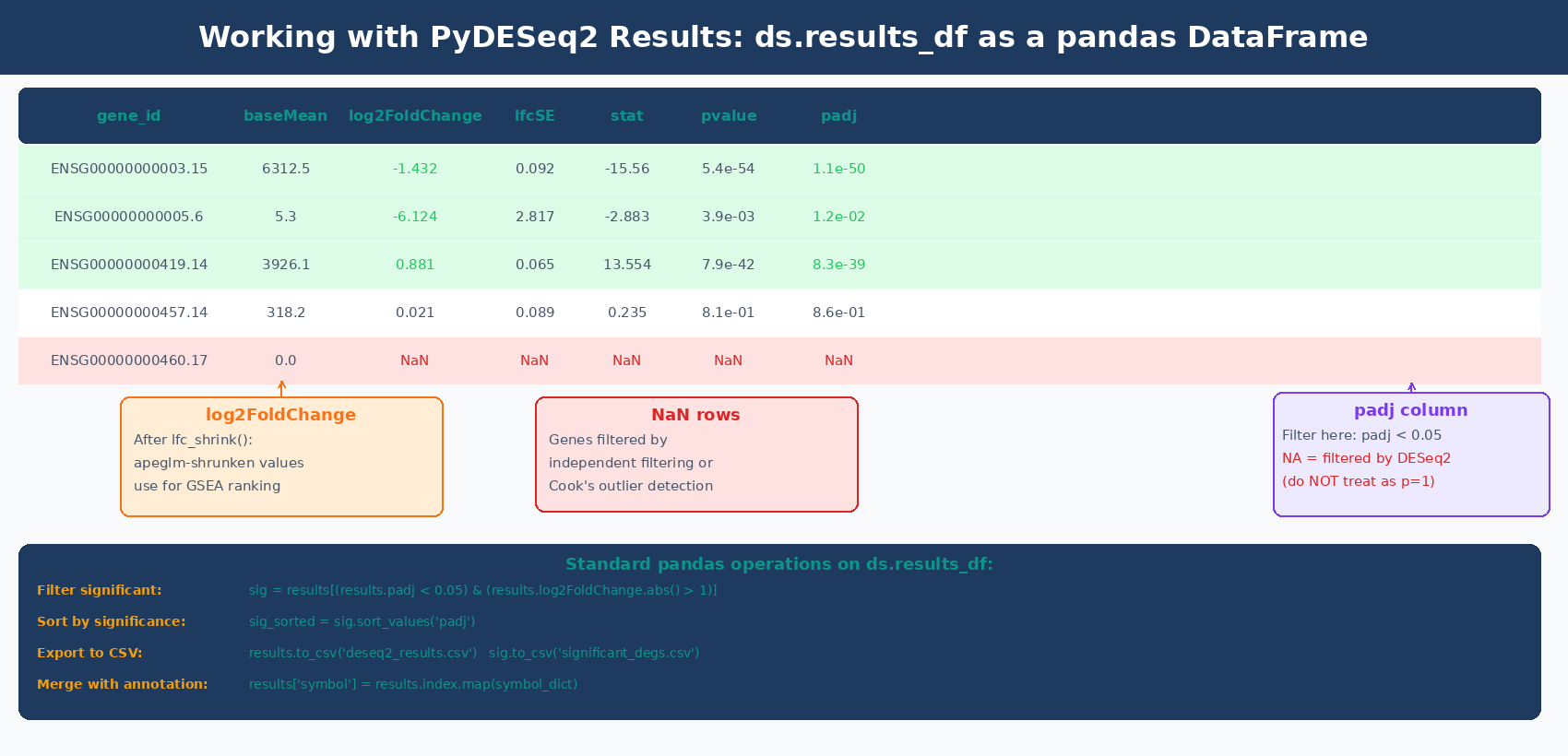
Running a Multi-Factor Design in PyDESeq2
Adding a batch variable or additional covariate to the design formula uses the same syntax as R:
# Multi-factor design: control for batch while testing conditiondds_batch = DeseqDataSet( counts = counts_df, metadata = metadata, design = "~batch + condition", # batch is modeled, condition is tested refit_cooks = True, inference = inference)dds_batch.deseq2()
# Stats: same as before — contrast specifies condition effectds_batch = DeseqStats( dds_batch, contrast = ["condition", "treated", "control"], alpha = 0.05, inference = inference)ds_batch.summary()ds_batch.lfc_shrink(coeff="condition_treated_vs_control")
results_batch = ds_batch.results_df.copy()The batch variable absorbs variance attributable to library preparation batches, which increases the power to detect the condition effect. If you know samples were prepared in batches, always include batch in the design.
Running Multiple Contrasts from One Fitted Model
When your experiment has three or more conditions, fit the model once and run multiple contrasts:
# Three-condition experiment: control, dose_low, dose_high# Assume metadata["condition"] has all three levels
dds_multi = DeseqDataSet( counts = counts_df_multi, metadata = metadata_multi, design = "~condition", inference = inference)dds_multi.deseq2()
# Run each contrast independently (model fitting is shared)contrasts_to_run = [ ("dose_low", "control"), ("dose_high", "control"), ("dose_high", "dose_low"),]
all_results = {}for tested, ref in contrasts_to_run: ds_c = DeseqStats( dds_multi, contrast = ["condition", tested, ref], alpha = 0.05, inference = inference ) ds_c.summary() ds_c.lfc_shrink(coeff=f"condition_{tested}_vs_{ref}") all_results[f"{tested}_vs_{ref}"] = ds_c.results_df.copy() n_sig = (ds_c.results_df["padj"] < 0.05).sum() print(f"{tested} vs {ref}: {n_sig} significant genes")Generating Visualizations from PyDESeq2 Results
PyDESeq2 includes a built-in MA plot method. For publication-quality plots, use matplotlib or seaborn on ds.results_df directly.
For a plotting deep dive, the same interpretation logic carries into How to Make Volcano Plots and MA Plots in R: ggplot2 and EnhancedVolcano, and the ranked-list follow-up is What Is GSEA and Why Does It Beat a Simple DEG List.
MA Plot
import matplotlib.pyplot as pltimport numpy as np
# Built-in MA plot (requires prior ds.summary() call)ds.plot_MA(s=5)plt.title("MA Plot: treated vs control")plt.savefig("results/ma_plot.pdf", bbox_inches="tight")Volcano Plot
fig, ax = plt.subplots(figsize=(8, 6))
# Compute -log10(padj), handle NaNplot_df = results.dropna(subset=["padj", "log2FoldChange"]).copy()plot_df["neg_log10_padj"] = -np.log10(plot_df["padj"].clip(lower=1e-300))
# Color by significanceis_sig = (plot_df["padj"] < 0.05) & (plot_df["log2FoldChange"].abs() > 1.0)
ax.scatter( plot_df.loc[~is_sig, "log2FoldChange"], plot_df.loc[~is_sig, "neg_log10_padj"], color = "lightgray", s = 5, alpha = 0.7, label = "Not significant")ax.scatter( plot_df.loc[is_sig, "log2FoldChange"], plot_df.loc[is_sig, "neg_log10_padj"], color = "#e63946", s = 8, alpha = 0.8, label = "Significant")
ax.axhline(-np.log10(0.05), ls="--", color="gray", lw=1)ax.axvline( 1.0, ls="--", color="gray", lw=1)ax.axvline(-1.0, ls="--", color="gray", lw=1)
ax.set_xlabel("log2 Fold Change (treated vs control)", fontsize=12)ax.set_ylabel("-log10(adjusted p-value)", fontsize=12)ax.set_title("Volcano Plot: treated vs control")ax.legend(frameon=False)
n_up = (is_sig & (plot_df["log2FoldChange"] > 0)).sum()n_down = (is_sig & (plot_df["log2FoldChange"] < 0)).sum()ax.text(0.98, 0.98, f"Up: {n_up}\nDown: {n_down}", transform=ax.transAxes, ha="right", va="top", fontsize=10)
plt.tight_layout()plt.savefig("results/volcano_plot.pdf", bbox_inches="tight", dpi=150)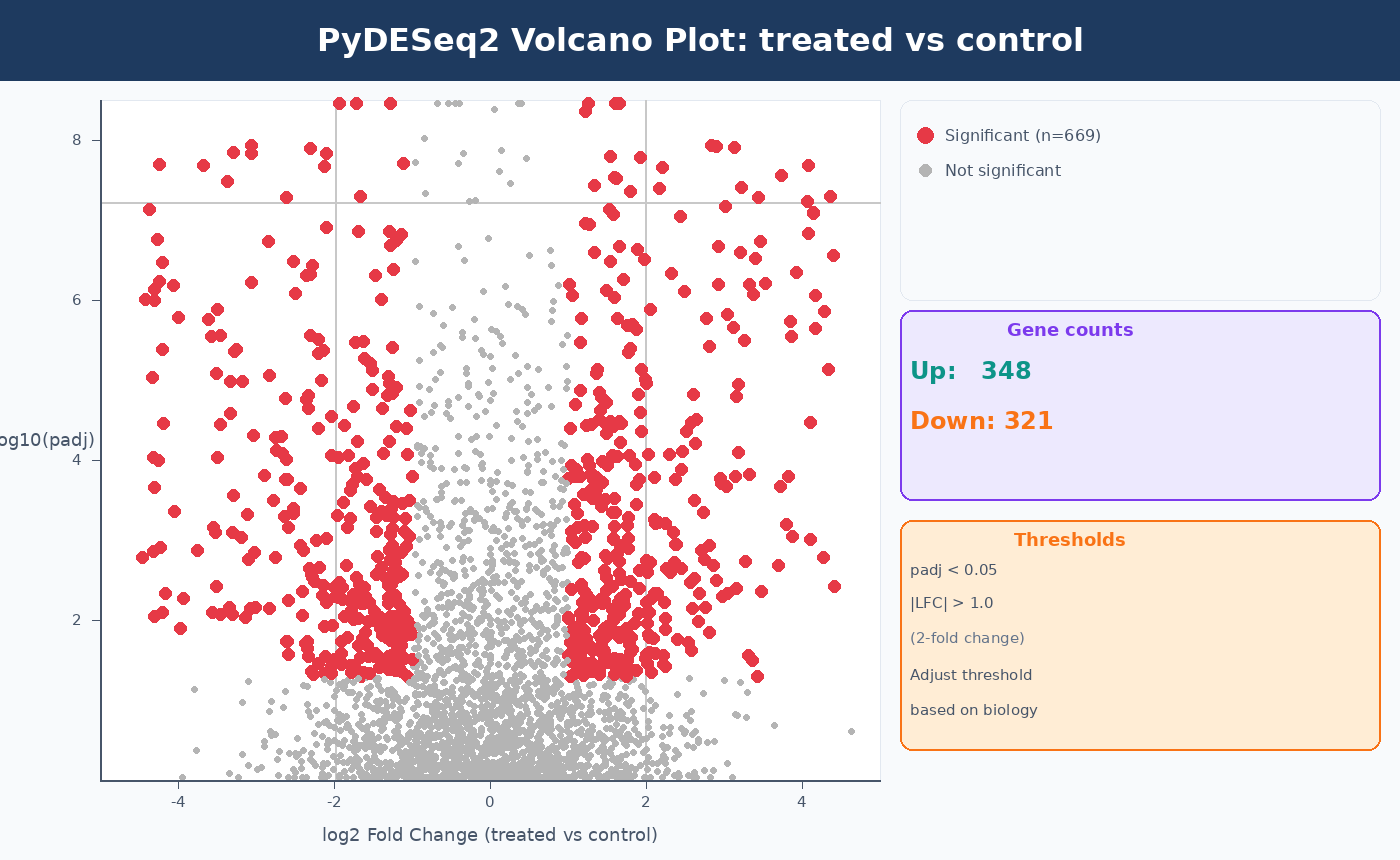
Complete PyDESeq2 Pipeline Script
Here is the full workflow in a single reusable script:
#!/usr/bin/env python3"""pydeseq2_pipeline.py
Complete PyDESeq2 differential expression analysis.Input: counts.csv (samples × genes, integer counts) metadata.csv (samples × variables)Output: pydeseq2_results_annotated.csv pydeseq2_significant_degs.csv volcano_plot.pdf"""
import pandas as pdimport numpy as npimport matplotlib.pyplot as pltfrom pydeseq2.dds import DeseqDataSetfrom pydeseq2.default_inference import DefaultInferencefrom pydeseq2.ds import DeseqStats
# ── 1. Load and validate inputs ──────────────────────────────────counts_raw = pd.read_csv("results/counts/counts.csv", index_col=0)counts_df = counts_raw.T.astype(int) # transpose: samples × genes
metadata = pd.read_csv("data/sample_metadata.csv", index_col=0, dtype=str) # keep all columns as strings
assert list(counts_df.index) == list(metadata.index), \ "Row order mismatch between counts and metadata"
print(f"Samples: {counts_df.shape[0]} Genes (pre-filter): {counts_df.shape[1]}")
# ── 2. Filter low-count genes ────────────────────────────────────genes_to_keep = counts_df.columns[counts_df.sum(axis=0) >= 10]counts_df = counts_df[genes_to_keep]print(f"Genes (post-filter): {counts_df.shape[1]}")
# ── 3. Fit model ─────────────────────────────────────────────────inference = DefaultInference(n_cpus=8)
dds = DeseqDataSet( counts = counts_df, metadata = metadata, design = "~condition", refit_cooks = True, inference = inference)dds.deseq2()
# ── 4. Run statistical test ──────────────────────────────────────ds = DeseqStats( dds, contrast = ["condition", "treated", "control"], alpha = 0.05, inference = inference)ds.summary()
# ── 5. Apply LFC shrinkage ───────────────────────────────────────results_unshrunk = ds.results_df.copy()ds.lfc_shrink(coeff="condition_treated_vs_control")results = ds.results_df.copy()
# ── 6. Filter and save ───────────────────────────────────────────sig = results[ (results["padj"] < 0.05) & (results["log2FoldChange"].abs() > 1.0)].sort_values("padj")
print(f"\nSignificant DEGs: {len(sig)}")print(f" Up: {(sig['log2FoldChange'] > 0).sum()} " f"Down: {(sig['log2FoldChange'] < 0).sum()}")
results.to_csv("results/pydeseq2_all_genes.csv")sig.to_csv("results/pydeseq2_significant_degs.csv")
# ── 7. Volcano plot ──────────────────────────────────────────────plot_df = results.dropna(subset=["padj"]).copy()plot_df["neg_log10_padj"] = -np.log10(plot_df["padj"].clip(lower=1e-300))is_sig = (plot_df["padj"] < 0.05) & (plot_df["log2FoldChange"].abs() > 1.0)
fig, ax = plt.subplots(figsize=(7, 5))ax.scatter(plot_df.loc[~is_sig, "log2FoldChange"], plot_df.loc[~is_sig, "neg_log10_padj"], s=4, color="lightgray", alpha=0.6)ax.scatter(plot_df.loc[is_sig, "log2FoldChange"], plot_df.loc[is_sig, "neg_log10_padj"], s=7, color="#e63946", alpha=0.8)ax.axhline(-np.log10(0.05), ls="--", color="gray", lw=0.8)ax.axvline(1.0, ls="--", color="gray", lw=0.8)ax.axvline(-1.0, ls="--", color="gray", lw=0.8)ax.set(xlabel="log2 Fold Change", ylabel="-log10(padj)", title="PyDESeq2: treated vs control")plt.tight_layout()plt.savefig("results/volcano_plot.pdf", dpi=150, bbox_inches="tight")print("Volcano plot saved.")NotchBio runs the equivalent of this pipeline internally for Python-based workflows, producing the same normalized counts, Wald test results, and shrunk LFCs in an automated environment. If you prefer to skip the environment setup and get directly to interpreting results, upload your count matrix and metadata at notchbio.app.
Further reading
Read another related post
How to Run DESeq2: A Complete Walkthrough from Count Matrix to Results
Step-by-step DESeq2 tutorial in R: build a DESeqDataSet, understand size factors and dispersion, run DESeq(), interpret results columns, apply lfcShrink with apeglm, and filter DEGs.
TutorialHow to Set Up a Bulk RNA-Seq Analysis Environment on Ubuntu and macOS
Step-by-step guide to installing Miniforge, conda, bioconda, R 4.4, and DESeq2 for bulk RNA-seq analysis. Reproducible environments, version pinning, and fixes for common install errors.
TutorialHow to Quantify RNA-Seq Reads with Salmon: Index, Quant, and Import to R
Step-by-step Salmon RNA-seq tutorial: build a decoy-aware index, run salmon quant on paired-end reads, understand quant.sf output, and import into DESeq2 with tximport.