Bulk RNA-Seq Is Not Dead: When To Use It Over scRNA-Seq
At a regional bioinformatics conference in 2025, a keynote speaker who had been working with RNA-seq since its early days made an observation that prompted knowing laughter from the audience: every talk that day had featured scRNA-seq data, which was ironic given that the technology was already, in his words, “old hat.” Spatial genomics was the new hotness, and the room could look forward to an identical conference three years from now dominated entirely by that.
The laughter was recognition. Single-cell RNA-seq has become so dominant in the literature and at conferences that researchers who publish bulk RNA-seq results sometimes feel they owe an apology for it. They do not. Bulk RNA-seq is not a legacy technology that laboratories are clinging to for sentimental reasons or budget constraints. For a large proportion of biological questions, it is the correct technology. The framing that positions single-cell as the default and bulk as the fallback has it backwards.
This post is about how to make the choice correctly, with a framework you can actually bring to a PI meeting.
The Hype-Reality Gap Is Measurable
The dominance of scRNA-seq in publications and conference programs does not reflect the distribution of experiments being run in most laboratories. Most groups sequencing RNA today are using bulk methods for most of their experiments. The reasons are not ignorance of single-cell technology; they are scientific appropriateness, cost, and statistical power.
A post on the r/bioinformatics community that garnered nearly 300 upvotes and 80 comments captured the frustration with conference saturation directly: every talk is just interpreting scRNA-seq data, every presentation another tSNE. The comments did not disagree with the premise. They pointed out that single-cell has become standard in the same way that bulk was standard a decade ago, which means it now appears everywhere regardless of whether it is the right tool for the question being asked.
That is the definition of hype: a technology applied beyond the domain where it genuinely outperforms alternatives. Understanding where single-cell legitimately wins, and where bulk legitimately wins, is the only way to make principled experimental design decisions rather than trend-following ones.
When Bulk RNA-Seq Is the Right Choice
Bulk RNA-seq aggregates signal across all cells in a sample, which is simultaneously its limitation and its strength depending on what you are trying to measure.
Statistical power with large cohorts. If your experiment involves comparing gene expression across dozens or hundreds of patient samples, population genetics datasets, or time-series experiments with many timepoints, bulk RNA-seq is the only practical option. Single-cell sequencing of 100 patient samples at 10,000 cells per sample is not a realistic budget or computational proposition for most groups. Bulk RNA-seq of 100 samples is standard. The statistical power that comes from large n is irreplaceable, and large n is where bulk operates without constraint.
Mechanism and pathway studies. When the biological question is “what pathways are activated when I apply treatment X to this cell line under condition Y,” and the cell line is reasonably homogeneous, bulk RNA-seq answers that question with high statistical sensitivity. You do not need single-cell resolution to detect that NF-kB signaling is upregulated in response to LPS. You need enough replicates and sufficient sequencing depth, both of which are far more achievable in bulk.
Regulatory and clinical contexts. RNA-seq data submitted to regulatory agencies, used in clinical trial analysis, or deposited in validated biomarker pipelines is almost always bulk. The analytical workflows are established, validated, and auditable. The statistical models are well understood. Single-cell data in regulatory contexts introduces interpretive complexity that most frameworks are not yet equipped to handle.
Longitudinal and intervention studies. Studies tracking gene expression changes over time in the same tissue, before and after drug treatment, or across disease progression stages benefit from the cost efficiency and throughput of bulk RNA-seq. Tracking 50 patient samples at five timepoints is a standard bulk experiment. The equivalent single-cell experiment would require careful cell type compositional analysis at every timepoint just to ensure you are comparing equivalent populations.
Total RNA and degraded samples. Single-cell RNA-seq requires intact, high-quality RNA from dissociated cells. Formalin-fixed paraffin-embedded tissue, archived samples, or any situation where RNA quality is compromised makes single-cell technically impractical. Bulk RNA-seq with appropriate library preparation handles these inputs routinely.
A rough cost comparison for budgeting
As of 2025, bulk RNA-seq library preparation and sequencing to 30 million reads costs roughly 150 to 300 USD per sample depending on facility and library prep kit. Single-cell RNA-seq with 10x Genomics to 5,000 cells per sample costs roughly 1,000 to 2,000 USD per sample including library prep and sequencing. For an experiment with 24 samples, that is approximately 4,000 to 7,000 USD for bulk versus 24,000 to 48,000 USD for single-cell. The single-cell experiment also requires substantially more computational resources and analysis time.
When Single-Cell RNA-Seq Is the Right Choice
Single-cell RNA-seq was developed to answer questions that bulk cannot, and those questions are real and important. The issue is not with the technology; it is with applying it to questions that do not require it.
Cellular heterogeneity and rare populations. If your tissue or sample contains multiple distinct cell types and you need to understand how each cell type responds to a perturbation, bulk RNA-seq averages over all of them. A treatment that strongly activates a rare immune cell population while leaving the majority epithelial cells unchanged will appear as a small, noisy signal in bulk data. Single-cell resolves this by attributing expression changes to specific cell populations.
Cell type identification and atlasing. Building a reference map of which cell types exist in a tissue, characterizing previously undescribed populations, or identifying marker genes that distinguish subtypes: these are questions that require single-cell resolution by definition. Bulk data cannot tell you which genes distinguish a macrophage from a dendritic cell in your specific tissue context.
Developmental trajectories and pseudotime analysis. Modeling continuous developmental transitions, ordering cells along a differentiation trajectory, or capturing the spectrum of states within a differentiating population requires single-cell data. Bulk RNA-seq captures a snapshot of average expression across the entire population and cannot reconstruct the ordering of intermediate states.
Clonal dynamics and mosaicism. Questions about how individual cell lineages behave differently from one another, or how somatic mutations are distributed across a tissue, require single-cell resolution that bulk cannot approximate.
The Deconvolution Middle Ground
Between true single-cell experiments and straightforward bulk experiments sits a practical middle ground: bulk RNA-seq with computational deconvolution.
If you have bulk RNA-seq data from a heterogeneous tissue and want to estimate cell type proportions across your samples, tools like CIBERSORTx and MuSiC can infer those proportions using a reference gene expression signature. This does not give you single-cell resolution, but it does give you an estimate of whether your treatment changed the relative abundance of immune cell populations, which is often the actual biological question.
Deconvolution works best when the reference signatures are tissue-matched and when the cell type composition differences between samples are relatively large. It breaks down when cell types are transcriptionally similar, when the reference does not cover all populations in your sample, or when you are trying to detect changes in rare populations at low frequency. In those cases, single-cell is genuinely required.
The practical decision rule is: if your question is about cell type proportions in a heterogeneous tissue and the expected compositional differences are large enough to detect by deconvolution, run bulk RNA-seq with deconvolution. If your question requires resolving transcriptional states within a cell type, or if the expected compositional differences are subtle, run single-cell.
A Decision Framework for Your PI Meeting
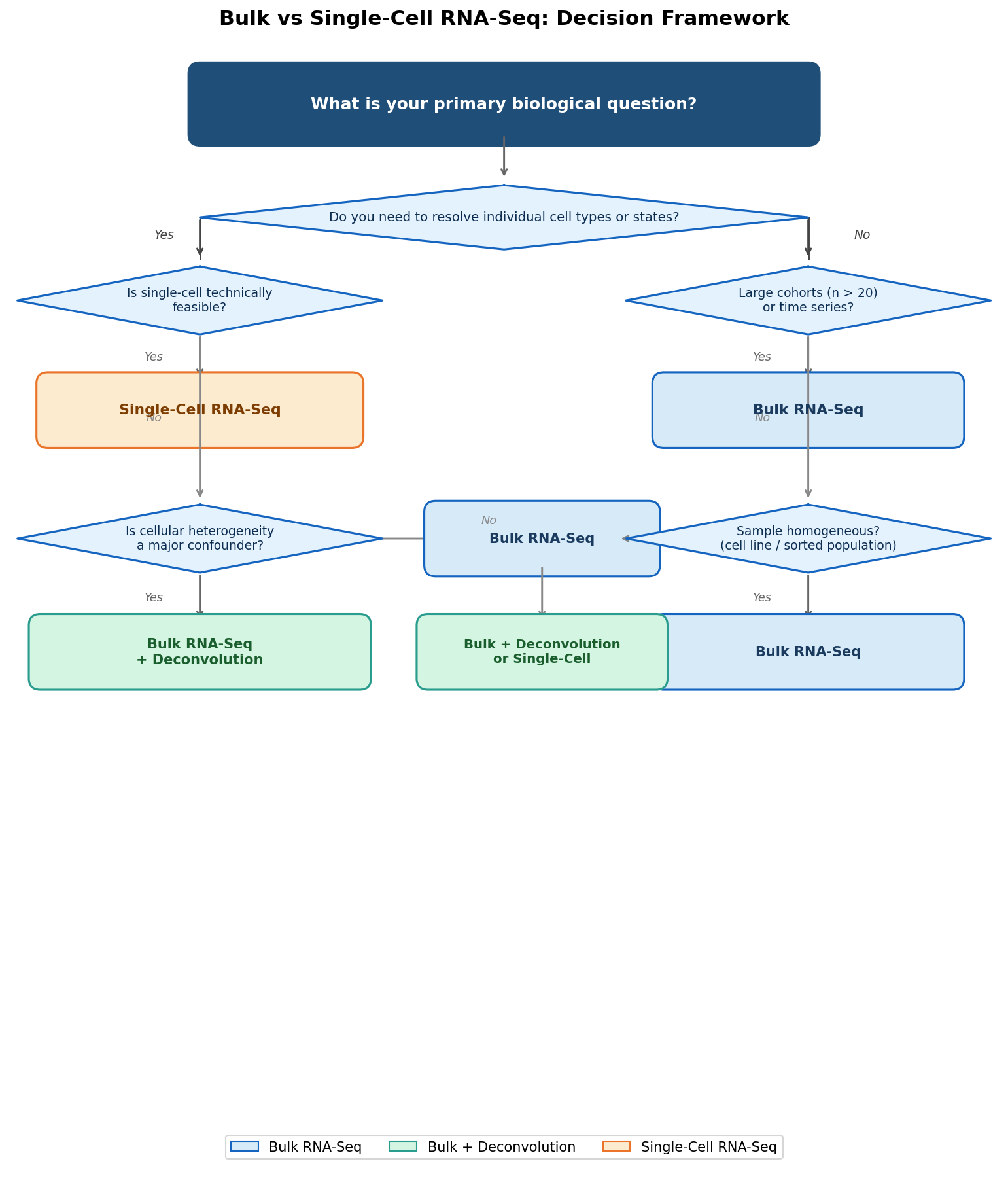
The Comparison Matrix
| Dimension | Bulk RNA-Seq | Bulk + Deconvolution | Single-Cell RNA-Seq |
|---|---|---|---|
| Cost per sample | Low (150 to 300 USD) | Low (same as bulk) | High (1,000 to 2,000 USD) |
| Cohort size feasibility | Large (100+) | Large (100+) | Small to medium (up to ~50) |
| Cell type resolution | None (averaged) | Estimated proportions | Full single-cell resolution |
| Rare cell type detection | Poor | Poor to moderate | Good |
| Trajectory analysis | Not possible | Not possible | Yes |
| Statistical power for DE | High with sufficient n | High with sufficient n | Lower per cell type |
| Regulatory acceptance | Established | Established | Emerging |
| Computational requirements | Moderate | Moderate to high | High |
| Works with degraded RNA | Yes | Yes | No |
| Analysis pipeline maturity | Very mature | Mature | Maturing rapidly |
Making the Decision Honestly
The right sequencing modality is the one that answers your biological question at the resolution it actually requires, within a budget and timeline that does not compromise your ability to achieve adequate statistical power.
Single-cell RNA-seq is remarkable technology. It has opened biological questions that bulk could not address and produced some of the most significant discoveries in cell biology of the past decade. It is also expensive, technically demanding, and analytically complex in ways that have enabled a substantial literature of underpowered, overinterpreted studies where single-cell was chosen because it looked impressive rather than because it was necessary.
Bulk RNA-seq, run correctly with sufficient replicates, appropriate statistical methods, and honest interpretation of the results, produces publishable, reproducible science at a fraction of the cost and computational overhead. The field’s tendency to treat it as a second-class option reflects conference culture more than scientific reality.
For most bulk RNA-seq workflows, NotchBio covers the full pipeline from FASTQ to differential expression and enrichment analysis, with validated defaults and reproducible run records. If bulk is the right choice for your experiment, the infrastructure to execute it well is not a bottleneck.
Related Reading
Further reading
Read another related post
How To Submit RNA-Seq Results That Reviewers Cannot Reject
Reviewers reject RNA-seq papers for predictable reasons: missing FDR correction, version-less methods, inaccessible data. A checklist that prevents it.
TutorialSalmon From FASTQ to Counts: A Complete Tutorial
A complete Salmon tutorial with decoy-aware indexing, quantification flags explained, tximport into R, DESeq2 integration, and QC checks at every step.
Research GuideHow To Write an RNA-Seq Methods Section Reviewers Accept
A reviewer-proof RNA-seq methods section is shorter than you think but far more specific. Templates, required elements, and what reviewers always flag missing.